From the Cardiovascular Medicine Division, Radcliffe Department of Medicine, University of Oxford, Oxford OX3 9DU, United Kingdom
From the Cardiovascular Medicine Division, Radcliffe Department of Medicine, University of Oxford, Oxford OX3 9DU, United Kingdom.
J Biol Chem. 2018 Jul 6;293(27):10487-10499. doi: 10.1074/jbc.RA118.002081. Epub 2018 May 14.
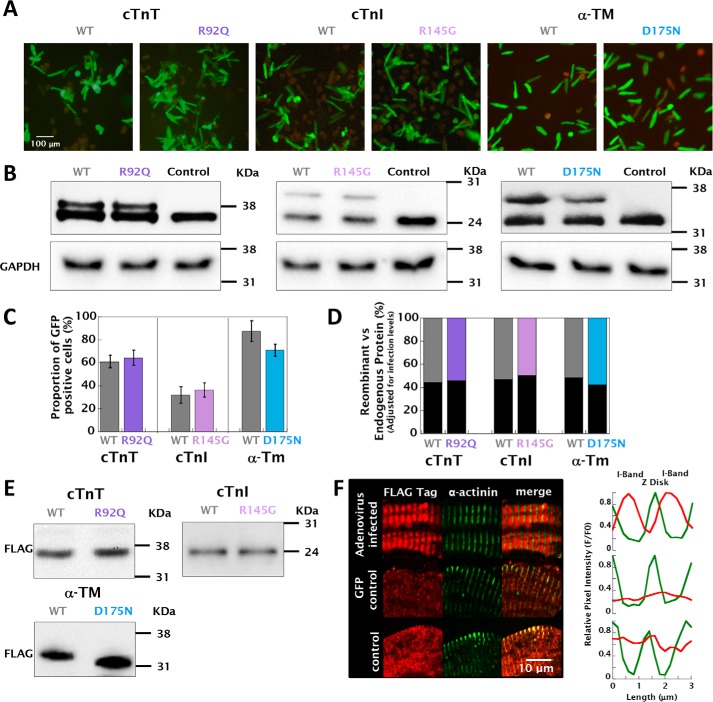
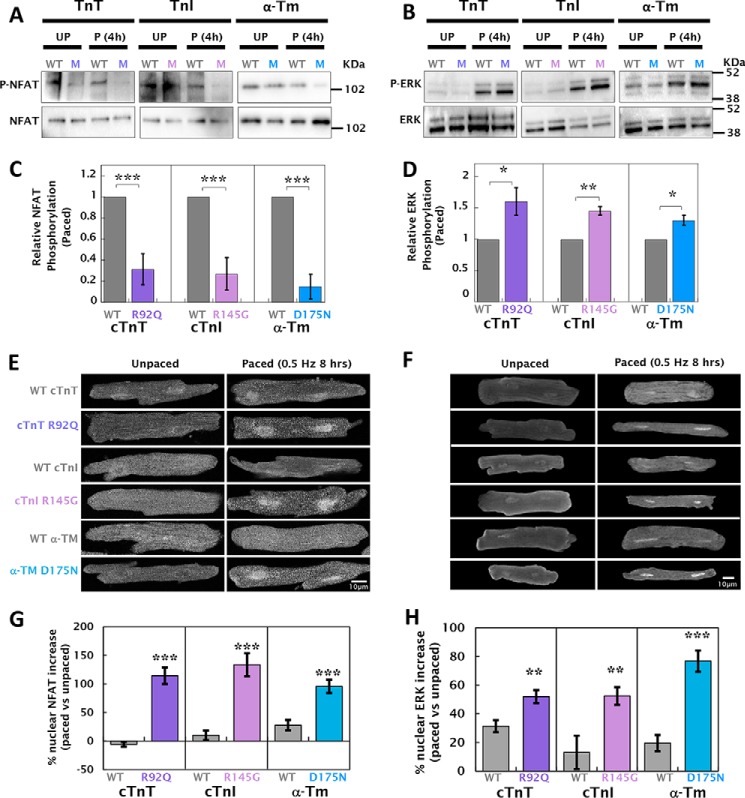
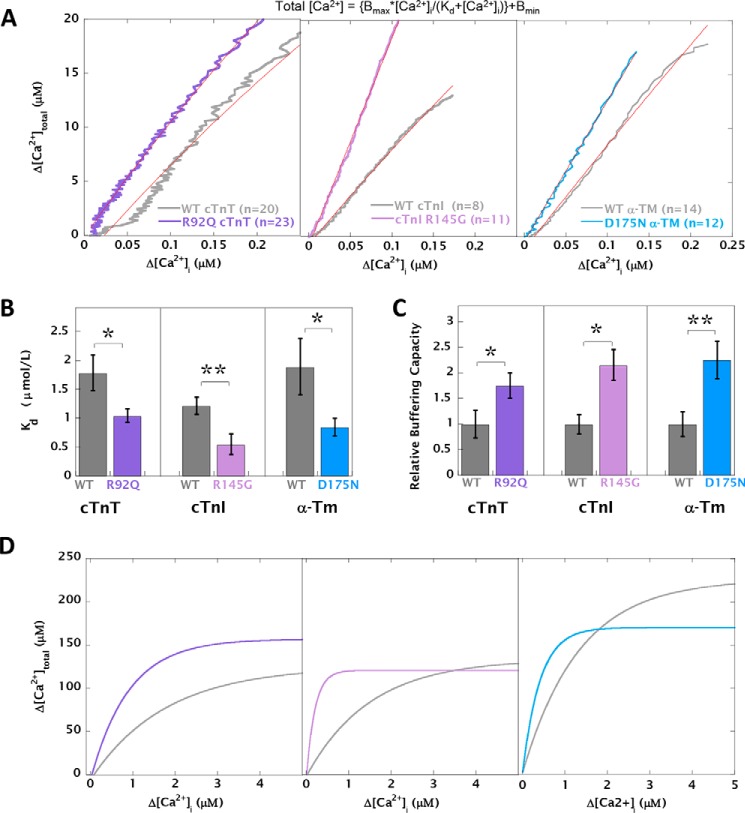
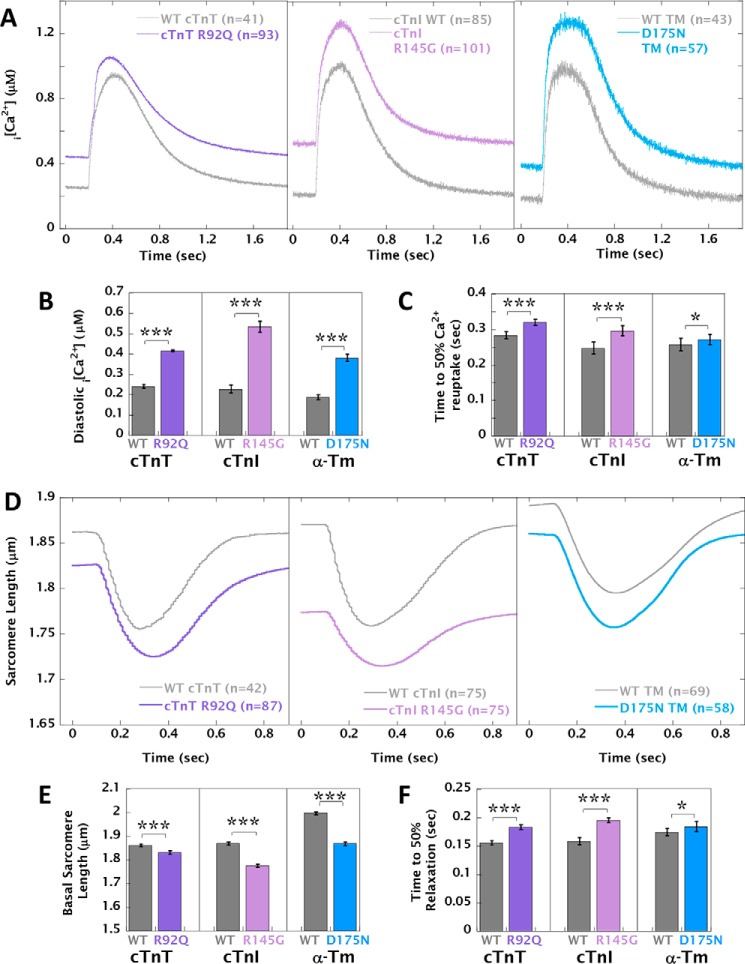
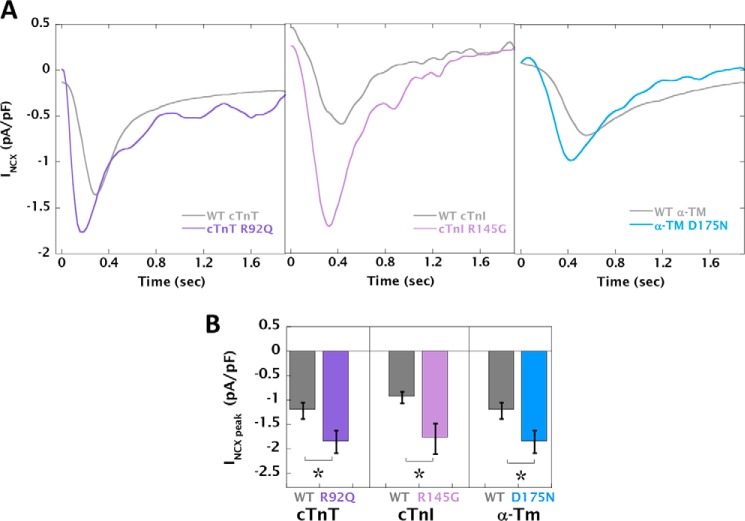
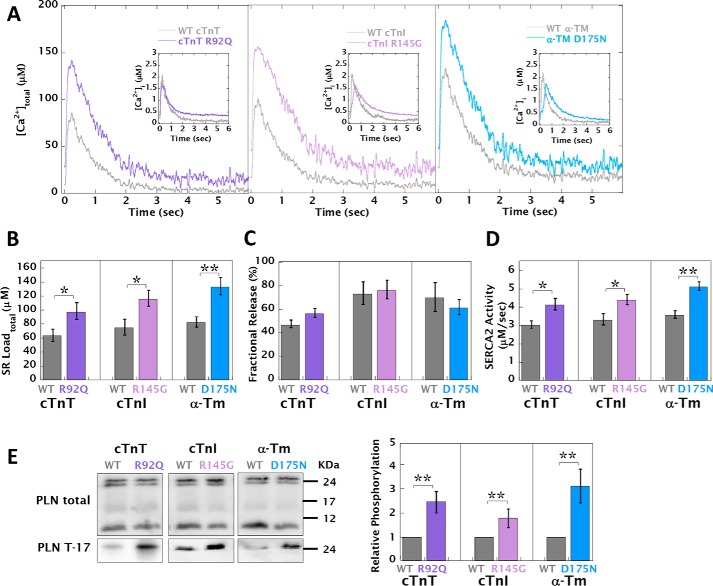
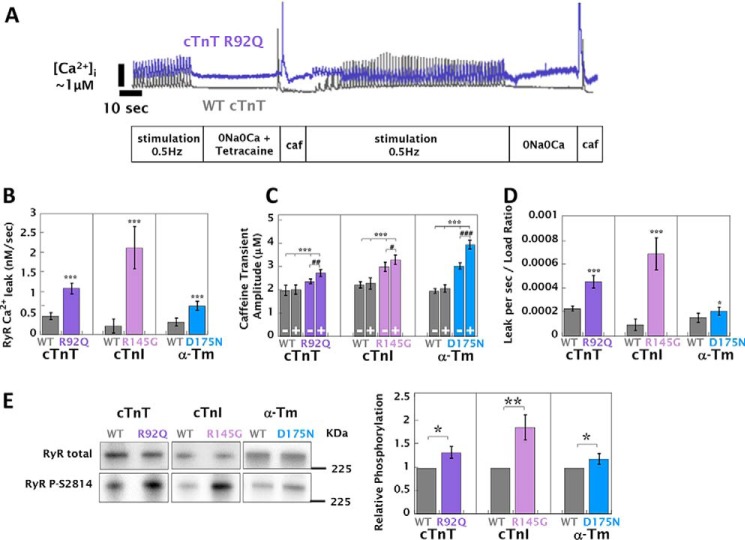
Mutations in thin filament regulatory proteins that cause hypertrophic cardiomyopathy (HCM) increase myofilament Ca sensitivity. Mouse models exhibit increased Ca buffering and arrhythmias, and we hypothesized that these changes are primary effects of the mutations (independent of compensatory changes) and that increased Ca buffering and altered Ca handling contribute to HCM pathogenesis via activation of Ca-dependent signaling. Here, we determined the primary effects of HCM mutations on intracellular Ca handling and Ca-dependent signaling in a model system possessing Ca-handling mechanisms and contractile protein isoforms closely mirroring the human environment in the absence of potentially confounding remodeling. Using adenovirus, we expressed HCM-causing variants of human troponin-T, troponin-I, and α-tropomyosin (R92Q, R145G, and D175N, respectively) in isolated guinea pig left ventricular cardiomyocytes. After 48 h, each variant had localized to the I-band and comprised ∼50% of the total protein. HCM mutations significantly lowered the of Ca binding, resulting in higher Ca buffering of mutant cardiomyocytes. We observed increased diastolic [Ca] and slowed Ca reuptake, coupled with a significant decrease in basal sarcomere length and slowed relaxation. HCM mutant cells had higher sodium/calcium exchanger activity, sarcoplasmic reticulum Ca load, and sarcoplasmic/endoplasmic reticulum calcium ATPase 2 (SERCA2) activity driven by Ca/calmodulin-dependent protein kinase II (CaMKII) phosphorylation of phospholamban. The ryanodine receptor (RyR) leak/load relationship was also increased, driven by CaMKII-mediated RyR phosphorylation. Altered Ca homeostasis also increased signaling via both calcineurin/NFAT and extracellular signal-regulated kinase pathways. Altered myofilament Ca buffering is the primary initiator of signaling cascades, indicating that directly targeting myofilament Ca sensitivity provides an attractive therapeutic approach in HCM.
导致肥厚型心肌病(HCM)的细丝调节蛋白突变会增加肌球蛋白丝 Ca 敏感性。小鼠模型表现出 Ca 缓冲增加和心律失常,我们假设这些变化是突变的主要影响(与代偿性变化无关),并且 Ca 缓冲增加和 Ca 处理改变通过激活 Ca 依赖性信号传导导致 HCM 发病机制。在这里,我们在缺乏潜在的重构混杂的情况下,使用具有 Ca 处理机制和收缩蛋白同工型的模型系统来确定 HCM 突变对细胞内 Ca 处理和 Ca 依赖性信号传导的主要影响,这些同工型与人类环境非常相似。我们使用腺病毒在分离的豚鼠左心室心肌细胞中表达了导致 HCM 的人类肌钙蛋白-T、肌钙蛋白-I 和α-原肌球蛋白的变体(分别为 R92Q、R145G 和 D175N)。48 小时后,每种变体都定位于 I 带,占总蛋白的约 50%。HCM 突变显著降低了 Ca 结合的,导致突变型心肌细胞的 Ca 缓冲能力增加。我们观察到舒张期 [Ca]增加和 Ca 再摄取速度减慢,同时伴有基础肌节长度显著降低和舒张速度减慢。HCM 突变细胞具有更高的钠/钙交换器活性、肌浆网 Ca 负荷和肌浆/内质网钙 ATP 酶 2(SERCA2)活性,这是由 Ca/钙调蛋白依赖性蛋白激酶 II(CaMKII)对肌球蛋白轻链磷酸化所驱动的。ryanodine 受体(RyR)的漏/载关系也增加了,这是由 CaMKII 介导的 RyR 磷酸化所驱动的。改变的 Ca 稳态也通过钙调神经磷酸酶/NFAT 和细胞外信号调节激酶途径增加信号。改变的肌球蛋白丝 Ca 缓冲是信号级联的主要启动子,表明直接靶向肌球蛋白丝 Ca 敏感性为 HCM 提供了一种有吸引力的治疗方法。