Wang Ying, Fu Lei, Ren Jie, Yu Zhaoxia, Chen Ting, Sun Fengzhu
Department of Automation, Xiamen University, Xiamen, China.
Molecular and Computational Biology Program, University of Southern California, Los Angeles, CA, United States.
Front Microbiol. 2018 May 3;9:872. doi: 10.3389/fmicb.2018.00872. eCollection 2018.
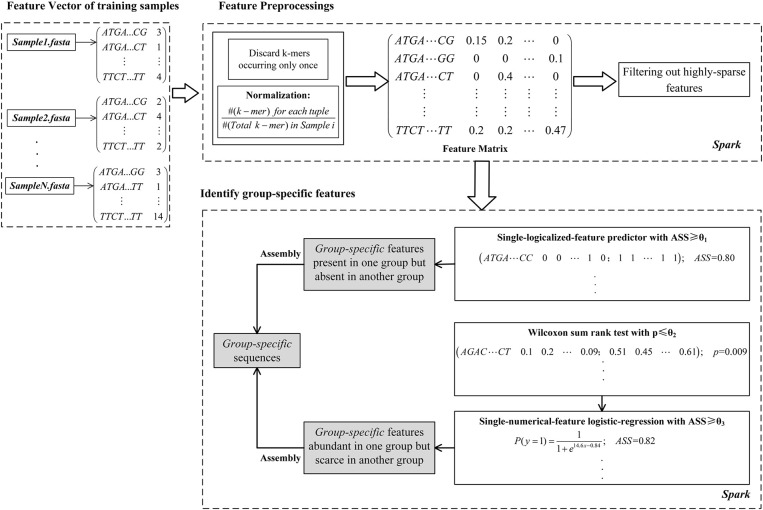
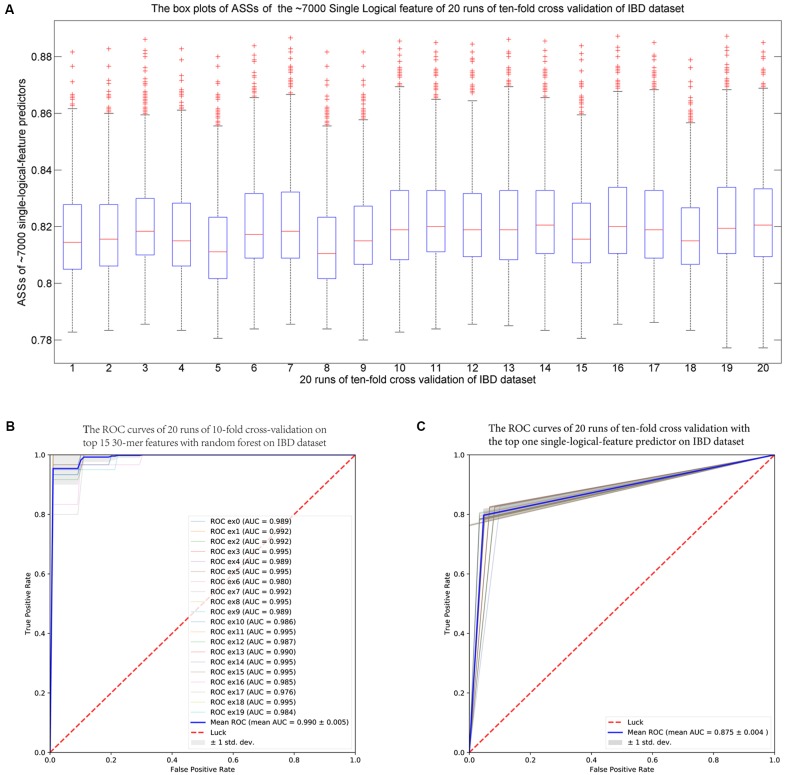
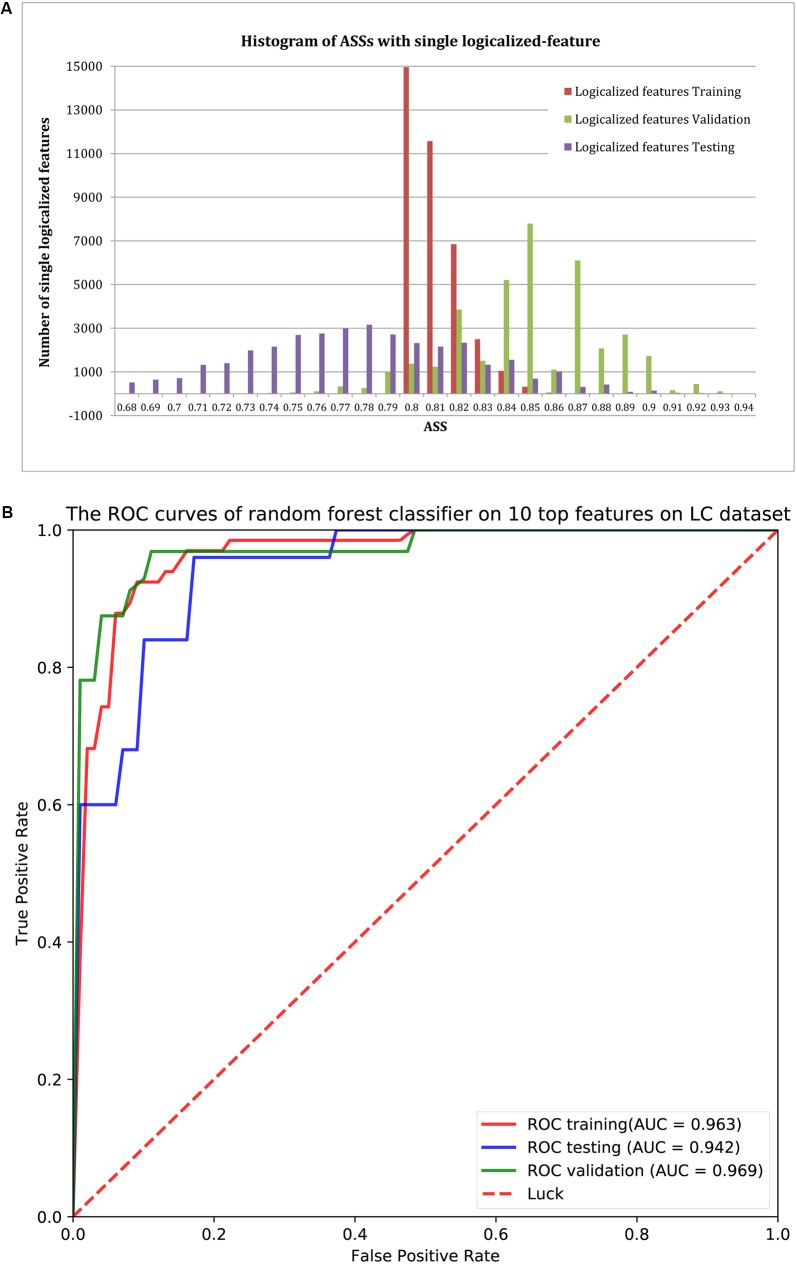
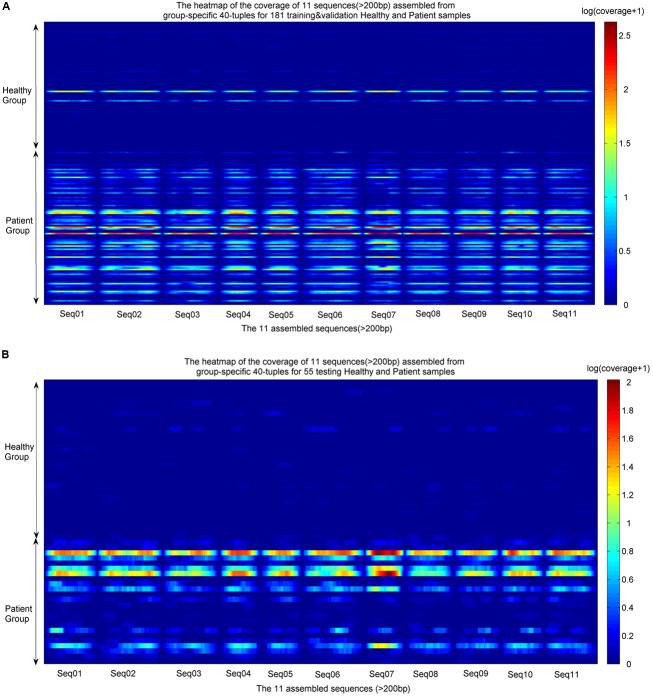
Comparing metagenomic samples is crucial for understanding microbial communities. For different groups of microbial communities, such as human gut metagenomic samples from patients with a certain disease and healthy controls, identifying sequences offers essential information for potential biomarker discovery. A sequence that is present, or rich, in one group, but absent, or scarce, in another group is considered "" in our study. Our main purpose is to discover sequence regions between control and case groups as disease-associated markers. We developed a long -mer ( ≥ 30 bps)-based computational pipeline to detect sequences at strain resolution free from reference sequences, sequence alignments, and metagenome-wide assembly. We called our method MetaGO: oligonucleotide analysis for metagenomic samples. An open-source pipeline on was developed with parallel computing. We applied MetaGO to one simulated and three real metagenomic datasets to evaluate the discriminative capability of identified markers. In the simulated dataset, 99.11% of logical -mers covered 98.89% regions from the disease-associated strain. In addition, 97.90% of numerical -mers covered 99.61 and 96.39% of differentially abundant genome and regions between two groups, respectively. For a large-scale metagenomic liver cirrhosis (LC)-associated dataset, we identified 37,647 mer features. Any one of the features can predict disease status of the training samples with the average of sensitivity and specificity higher than 0.8. The random forests classification using the top 10 features yielded a higher AUC (from ∼0.8 to ∼0.9) than that of previous studies. All mers were present in LC patients, but not healthy controls. All the assembled 11 sequences can be mapped to two strains of : UTDB1-3 and DSM2008. The experiments on the other two real datasets related to Inflammatory Bowel Disease and Type 2 Diabetes in Women consistently demonstrated that MetaGO achieved better prediction accuracy with fewer features compared to previous studies. The experiments showed that MetaGO is a powerful tool for identifying -mers, which would be clinically applicable for disease prediction. MetaGO is available at https://github.com/VVsmileyx/MetaGO.
比较宏基因组样本对于理解微生物群落至关重要。对于不同的微生物群落组,例如来自患有某种疾病的患者和健康对照的人类肠道宏基因组样本,识别序列为潜在生物标志物的发现提供了重要信息。在我们的研究中,在一组中存在或丰富而在另一组中不存在或稀少的序列被视为“……”。我们的主要目的是发现对照组和病例组之间的序列区域作为疾病相关标志物。我们开发了一种基于长寡聚体(≥30个碱基对)的计算流程,以在无需参考序列、序列比对和全宏基因组组装的情况下,以菌株分辨率检测序列。我们将我们的方法称为MetaGO:宏基因组样本的寡核苷酸分析。利用并行计算在……上开发了一个开源流程。我们将MetaGO应用于一个模拟和三个真实宏基因组数据集,以评估所识别的……标志物的判别能力。在模拟数据集中,99.11%的……逻辑寡聚体覆盖了来自疾病相关菌株的98.89%的……区域。此外,97.90%的……数值寡聚体分别覆盖了两组之间差异丰富的基因组和区域的99.61%和96.39%。对于一个大规模的与肝硬化(LC)相关的宏基因组数据集,我们识别出37,647个寡聚体特征。任何一个特征都可以预测训练样本的疾病状态,敏感性和特异性的平均值高于0.8。使用前10个……特征的随机森林分类产生的AUC(从约0.8到约0.9)高于先前的研究。所有寡聚体均存在于LC患者中,但在健康对照中不存在。所有组装的11个……序列都可以映射到两种……菌株:UTDB1 - 3和DSM2008。在另外两个与炎症性肠病和女性2型糖尿病相关的真实数据集上的实验一致表明,与先前的研究相比,MetaGO以更少的特征实现了更好的预测准确性。实验表明,MetaGO是识别……寡聚体的强大工具,在临床上可用于疾病预测。可在https://github.com/VVsmileyx/MetaGO获取MetaGO。