Sun Qi, Peng Yifan, Liu Jinze
Department of Computer Science, University of Kentucky, Lexington, KY, 40508, USA.
Department of Population Health Sciences, Weill Cornell Medicine, New York, NY 10065, USA.
iScience. 2021 Jul 14;24(8):102855. doi: 10.1016/j.isci.2021.102855. eCollection 2021 Aug 20.
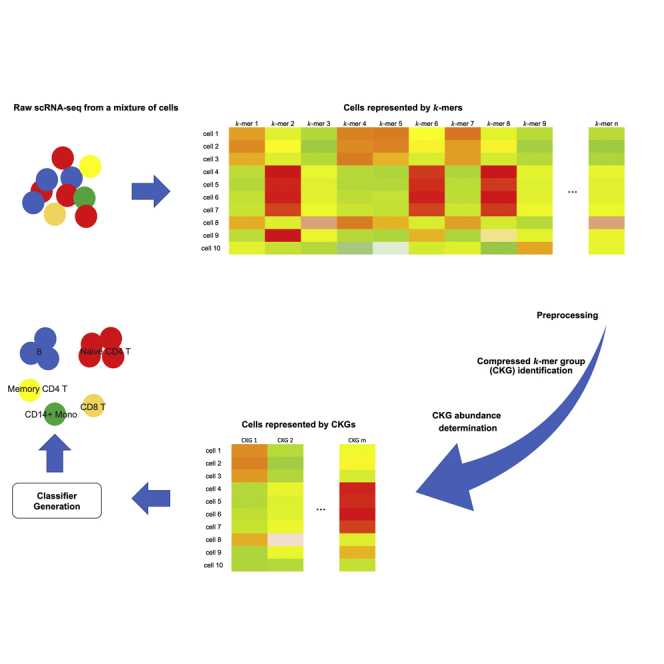
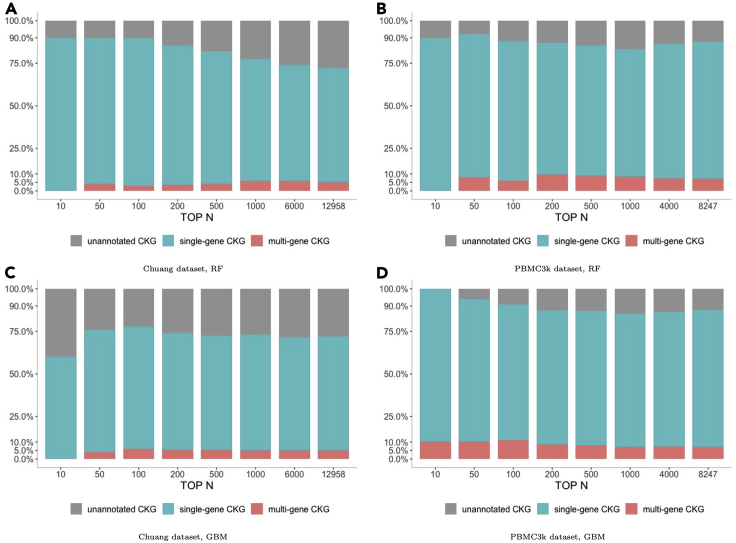
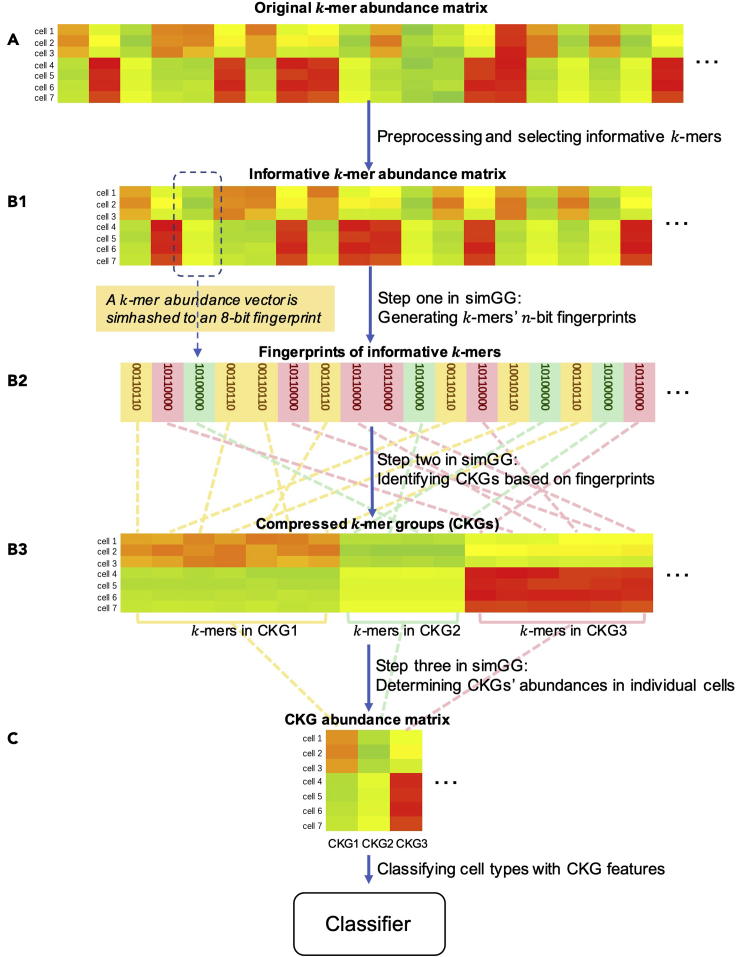
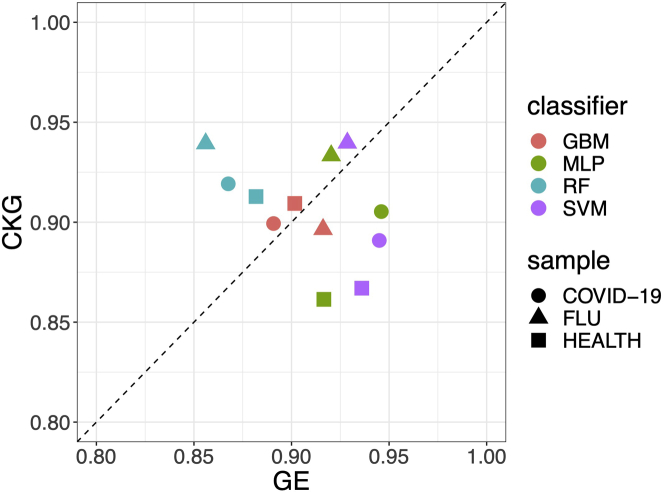
Single-cell RNA sequencing (scRNA-seq) has become a revolutionary technology to characterize cells under different biological conditions. Unlike bulk RNA-seq, gene expression from scRNA-seq is highly sparse due to limited sequencing depth per cell. This is worsened by tossing away a significant portion of reads that attribute to gene quantification. To overcome data sparsity and fully utilize original reads, we propose scSimClassify, a reference-free and alignment-free approach to classify cell types with -mer level features. The compressed -mer groups (CKGs), identified by the simhash method, contain -mers with similar abundance profiles and serve as the cells' features. Our experiments demonstrate that CKG features lend themselves to better performance than gene expression features in scRNA-seq classification accuracy in the majority of experimental cases. Because CKGs are derived from raw reads without alignment to reference genome, scSimClassify offers an effective alternative to existing methods especially when reference genome is incomplete or insufficient to represent subject genomes.
单细胞RNA测序(scRNA-seq)已成为一种革命性技术,用于表征不同生物学条件下的细胞。与批量RNA测序不同,由于每个细胞的测序深度有限,scRNA-seq的基因表达非常稀疏。通过舍弃大量用于基因定量的读数,这种情况会变得更糟。为了克服数据稀疏性并充分利用原始读数,我们提出了scSimClassify,这是一种无参考和无比对的方法,用于使用-mer水平特征对细胞类型进行分类。通过simhash方法识别的压缩-mer组(CKG)包含具有相似丰度谱的-mer,并作为细胞的特征。我们的实验表明,在大多数实验情况下,CKG特征在scRNA-seq分类准确性方面比基因表达特征具有更好的性能。由于CKG是从原始读数中衍生而来,无需与参考基因组比对,scSimClassify为现有方法提供了一种有效的替代方案,特别是当参考基因组不完整或不足以代表目标基因组时。