Department of Cellular and Molecular Medicine, University of California, San Diego, San Diego, USA.
Division of Evolutionary Biology, Faculty of Biology, Ludwig-Maximilian Universität München, Planegg-Martinsried, Germany.
Nucleic Acids Res. 2018 Aug 21;46(14):7006-7021. doi: 10.1093/nar/gky491.
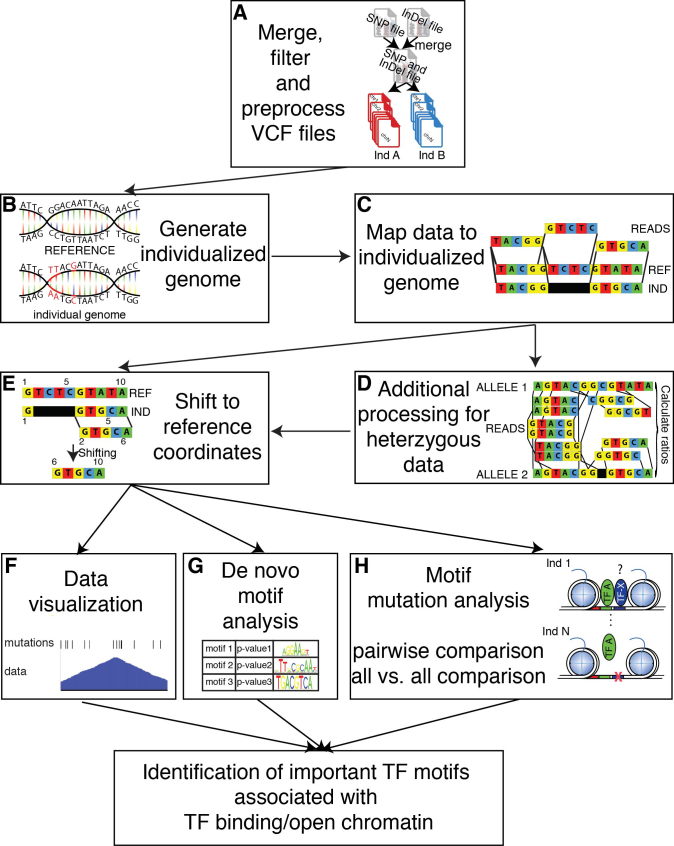
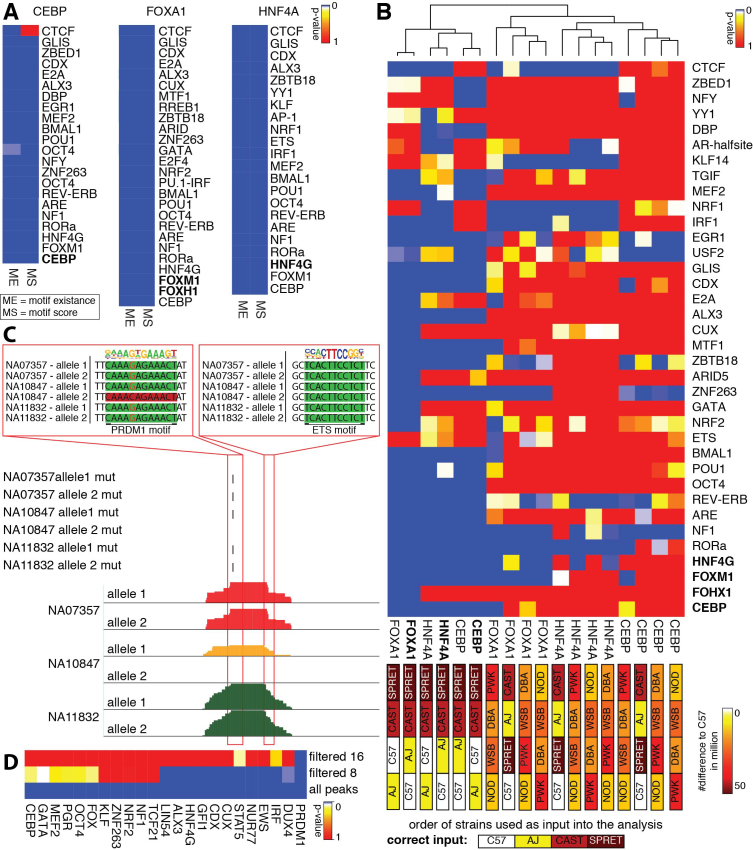
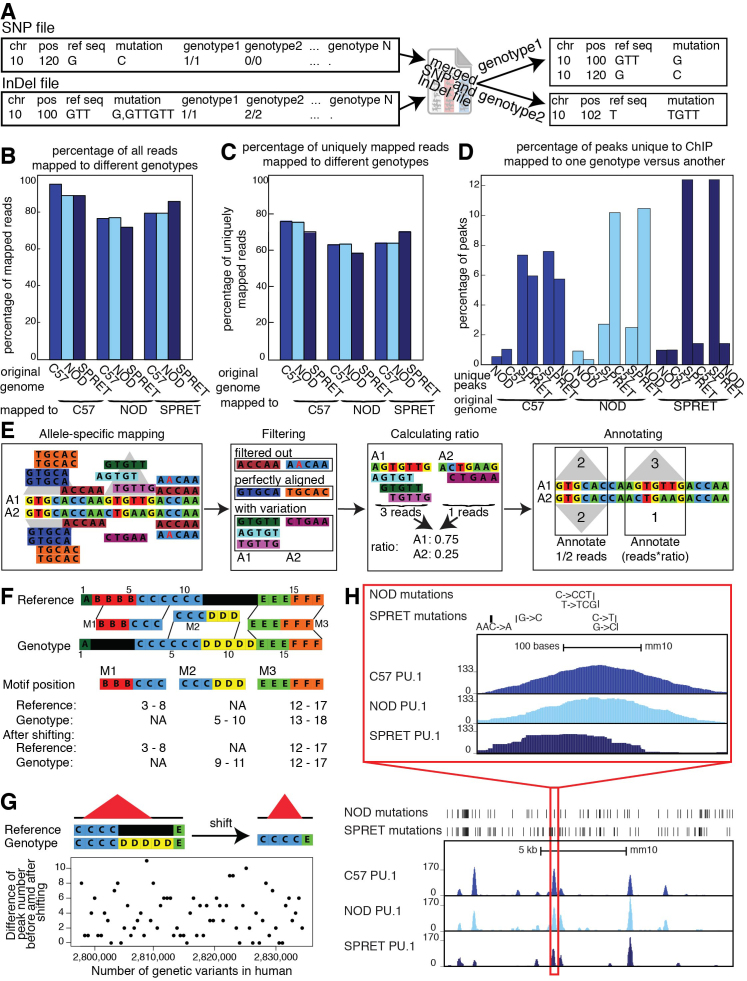
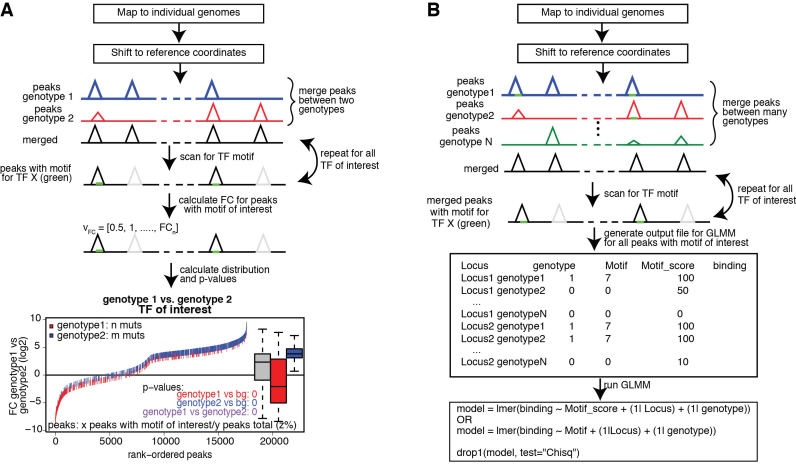
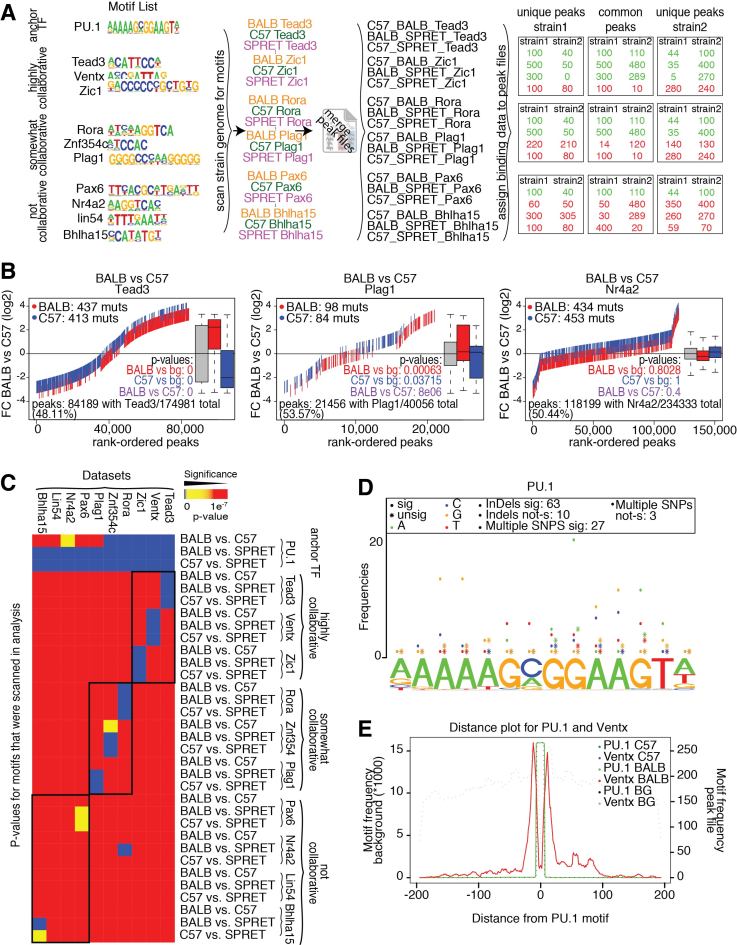
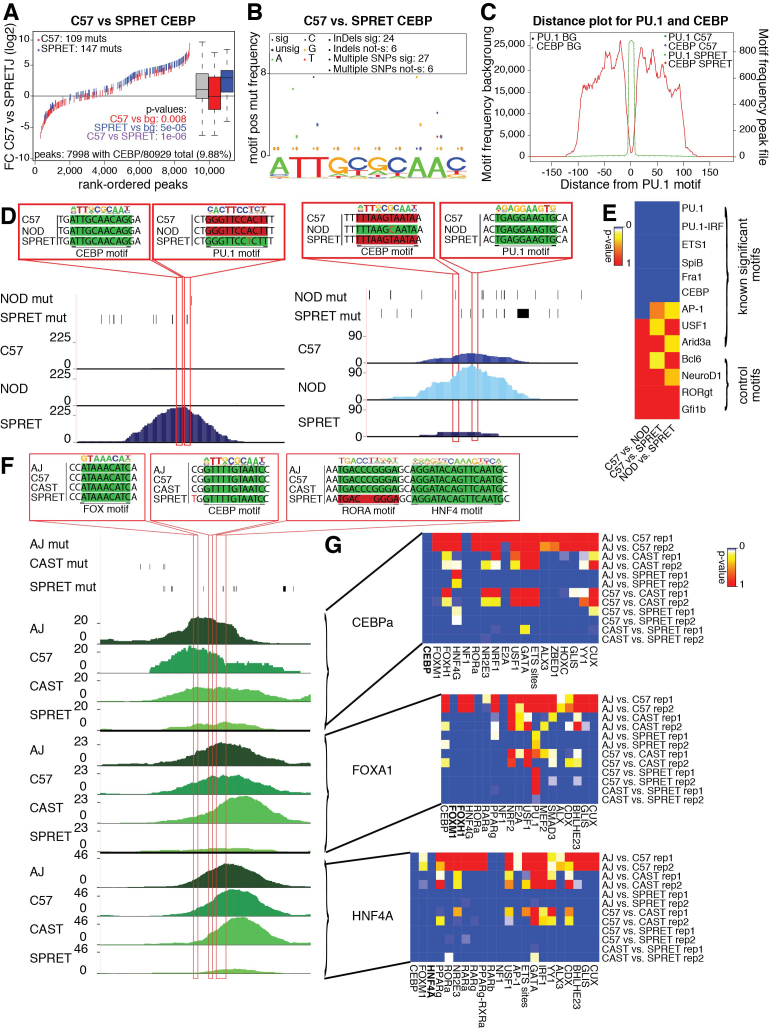
Cell-specific patterns of gene expression are determined by combinatorial actions of sequence-specific transcription factors at cis-regulatory elements. Studies indicate that relatively simple combinations of lineage-determining transcription factors (LDTFs) play dominant roles in the selection of enhancers that establish cell identities and functions. LDTFs require collaborative interactions with additional transcription factors to mediate enhancer function, but the identities of these factors are often unknown. We have shown that natural genetic variation between individuals has great utility for discovering collaborative transcription factors. Here, we introduce MMARGE (Motif Mutation Analysis of Regulatory Genomic Elements), the first publicly available suite of software tools that integrates genome-wide genetic variation with epigenetic data to identify collaborative transcription factor pairs. MMARGE is optimized to work with chromatin accessibility assays (such as ATAC-seq or DNase I hypersensitivity), as well as transcription factor binding data collected by ChIP-seq. Herein, we provide investigators with rationale for each step in the MMARGE pipeline and key differences for analysis of datasets with different experimental designs. We demonstrate the utility of MMARGE using mouse peritoneal macrophages, liver cells, and human lymphoblastoid cells. MMARGE provides a powerful tool to identify combinations of cell type-specific transcription factors while simultaneously interpreting functional effects of non-coding genetic variation.
基因表达的细胞特异性模式是由顺式调控元件中序列特异性转录因子的组合作用决定的。研究表明,相对简单的谱系决定转录因子(LDTFs)组合在选择建立细胞身份和功能的增强子方面起着主导作用。LDTFs 需要与其他转录因子进行协作相互作用来介导增强子功能,但这些因子的身份通常是未知的。我们已经表明,个体之间的自然遗传变异在发现协作转录因子方面具有很大的用处。在这里,我们介绍了 MMARGE(调控基因组元件的基序突变分析),这是第一个公开的软件工具套件,它将全基因组遗传变异与表观遗传数据集成在一起,以识别协作转录因子对。MMARGE 经过优化,可与染色质可及性测定(如 ATAC-seq 或 DNase I 超敏性)以及通过 ChIP-seq 收集的转录因子结合数据一起使用。在此,我们为 MMARGE 管道的每个步骤以及用于分析具有不同实验设计的数据集的关键差异提供了研究人员的基本原理。我们使用小鼠腹膜巨噬细胞、肝细胞和人淋巴母细胞样细胞演示了 MMARGE 的实用性。MMARGE 提供了一种强大的工具,可以识别细胞类型特异性转录因子的组合,同时解释非编码遗传变异的功能影响。