Molecular Microbiology and Structural Biochemistry, Unité Mixte de Recherche, Université Claude Bernard Lyon 1, Centre National de la Recherche Scientifique, 69367 Lyon Cedex 07, France.
Nucleic Acids Res. 2018 Jul 2;46(W1):W417-W422. doi: 10.1093/nar/gky472.
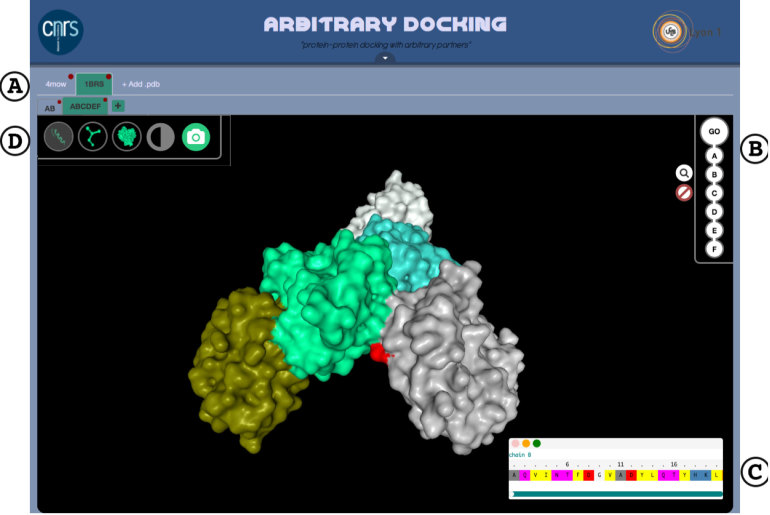
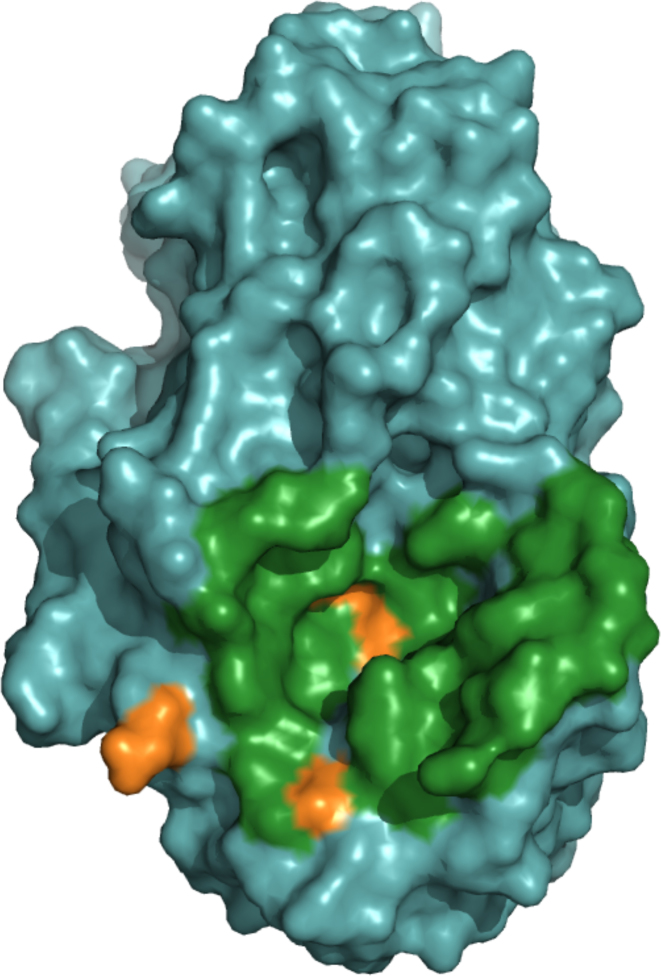
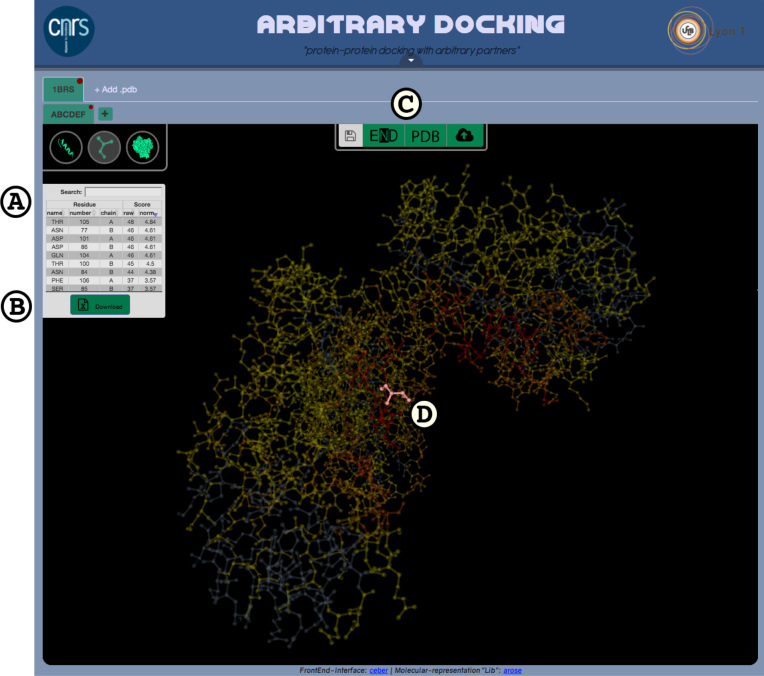
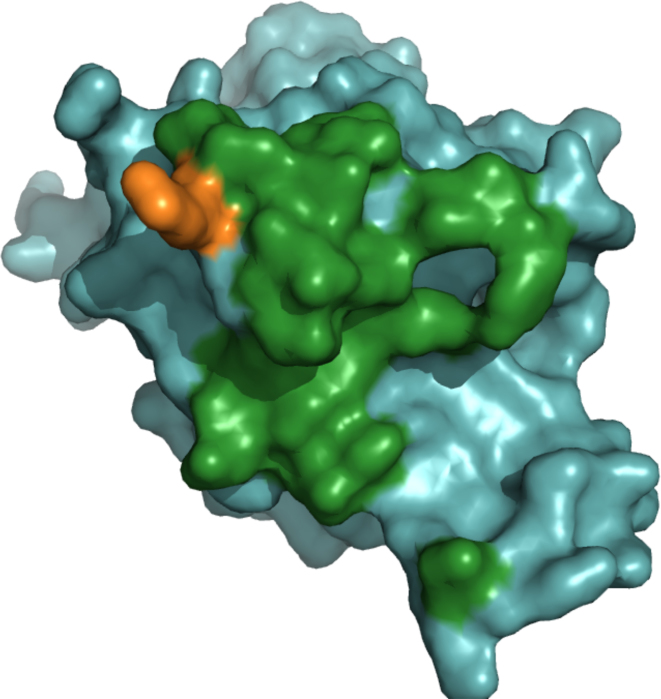
ArDock (ardock.ibcp.fr) is a structural bioinformatics web server for the prediction and the visualization of potential interaction regions at protein surfaces. ArDock ranks the surface residues of a protein according to their tendency to form interfaces in a set of predefined docking experiments between the query protein and a set of arbitrary protein probes. The ArDock methodology is derived from large scale cross-docking studies where it was observed that randomly chosen proteins tend to dock in a non-random way at protein surfaces. The method predicts interaction site of the protein, or alternate interfaces in the case of proteins with multiple interaction modes. The server takes a protein structure as input and computes a score for each surface residue. Its output focuses on the interactive visualization of results and on interoperability with other services.
ArDock(ardock.ibcp.fr)是一个结构生物信息学网络服务器,用于预测和可视化蛋白质表面的潜在相互作用区域。ArDock 根据查询蛋白质与一组任意蛋白质探针之间的一组预定义对接实验中形成界面的倾向,对蛋白质的表面残基进行排序。ArDock 方法源自大规模的交叉对接研究,在这些研究中观察到,随机选择的蛋白质往往以非随机的方式在蛋白质表面对接。该方法预测蛋白质的相互作用位点,或者在具有多种相互作用模式的蛋白质的情况下预测替代界面。该服务器以蛋白质结构作为输入,并为每个表面残基计算一个分数。其输出侧重于结果的交互式可视化以及与其他服务的互操作性。