Poltavsky Igor, Tkatchenko Alexandre
Fritz-Haber-Institut der Max-Planck-Gesellschaft , Faradayweg 4-6 , 14195 Berlin , Germany . Email:
Chem Sci. 2016 Feb 1;7(2):1368-1372. doi: 10.1039/c5sc03443d. Epub 2015 Oct 30.
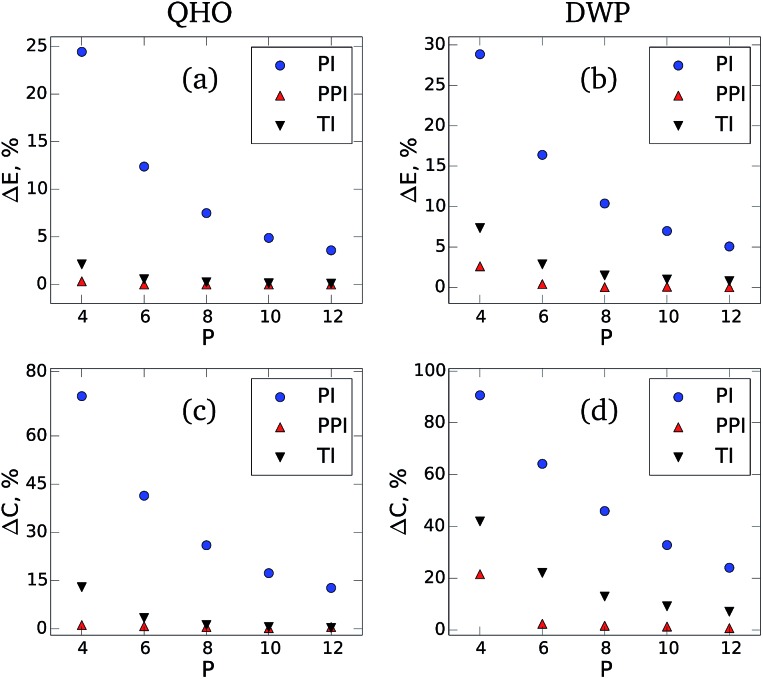
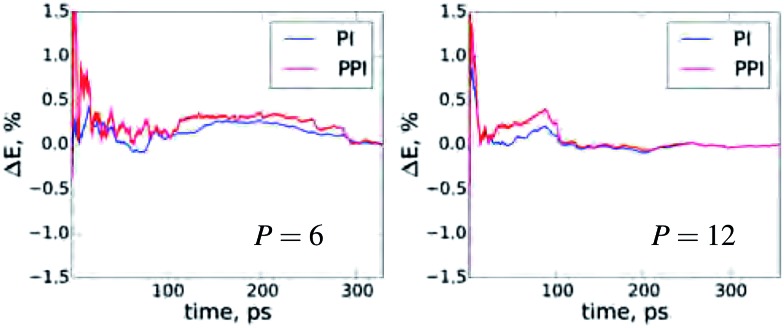
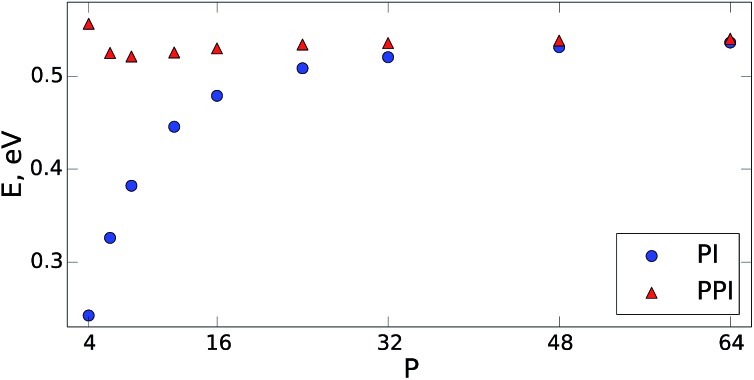
The quantum nature of nuclear motions plays a vital role in the structure, stability, and thermodynamics of molecules and materials. The standard approach to model nuclear quantum fluctuations in chemical and biological systems is to use path-integral molecular dynamics. Unfortunately, conventional path-integral simulations can have an exceedingly large computational cost due to the need to employ an excessive number of coupled classical subsystems (beads) for quantitative accuracy. Here, we combine perturbation theory with the Feynman-Kac imaginary-time path integral approach to quantum mechanics and derive an improved non-empirical partition function and estimators to calculate converged quantum observables. Our perturbed path-integral (PPI) method requires the same ingredients as the conventional approach, but increases the accuracy and efficiency of path integral simulations by an order of magnitude. Results are presented for the thermodynamics of fundamental model systems, an empirical water model containing 256 water molecules within periodic boundary conditions, and simulations of nitrogen and benzene molecules. For all of these examples, PPI simulations with 4 to 8 classical beads recover the nuclear quantum contribution to the total energy and heat capacity at room temperature within a 3% accuracy, paving the way toward seamless modeling of nuclear quantum effects in realistic molecules and materials.
核运动的量子本质在分子和材料的结构、稳定性及热力学中起着至关重要的作用。在化学和生物系统中模拟核量子涨落的标准方法是使用路径积分分子动力学。不幸的是,由于需要为保证定量精度而采用过多耦合的经典子系统(珠子),传统的路径积分模拟可能会有极高的计算成本。在此,我们将微扰理论与量子力学的费曼 - 卡茨虚时路径积分方法相结合,推导出一种改进的非经验配分函数和估计量,以计算收敛的量子可观测量。我们的微扰路径积分(PPI)方法所需的要素与传统方法相同,但将路径积分模拟的精度和效率提高了一个数量级。文中给出了基本模型系统的热力学结果、在周期性边界条件下包含256个水分子的经验水模型,以及氮气和苯分子的模拟结果。对于所有这些例子,使用4至8个经典珠子的PPI模拟在室温下能以3%的精度恢复核量子对总能量和热容的贡献,为在实际分子和材料中无缝模拟核量子效应铺平了道路。