Cobb-Vantress Inc., 4703 US HWY 412 E, Siloam Springs, AR, 72761, USA.
Department of Animal Science, Iowa State University, Ames, IA, 50010, USA.
Genet Sel Evol. 2018 Jun 19;50(1):32. doi: 10.1186/s12711-018-0402-1.
Population stratification and cryptic relationships have been the main sources of excessive false-positives and false-negatives in population-based association studies. Many methods have been developed to model these confounding factors and minimize their impact on the results of genome-wide association studies. In most of these methods, a two-stage approach is applied where: (1) methods are used to determine if there is a population structure in the sample dataset and (2) the effects of population structure are corrected either by modeling it or by running a separate analysis within each sub-population. The objective of this study was to evaluate the impact of population structure on the accuracy and power of genome-wide association studies using a Bayesian multiple regression method.
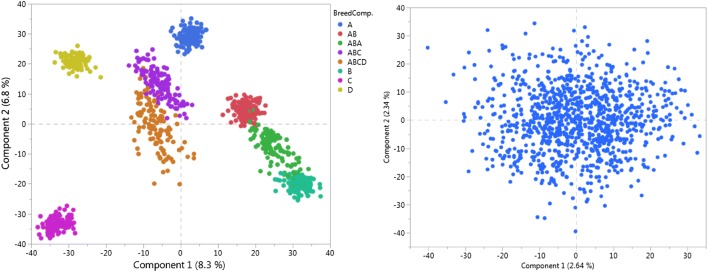
We conducted a genome-wide association study in a stochastically simulated admixed population. The genome was composed of six chromosomes, each with 1000 markers. Fifteen segregating quantitative trait loci contributed to the genetic variation of a quantitative trait with heritability of 0.30. The impact of genetic relationships and breed composition (BC) on three analysis methods were evaluated: single marker simple regression (SMR), single marker mixed linear model (MLM) and Bayesian multiple-regression analysis (BMR). Each method was fitted with and without BC. Accuracy, power, false-positive rate and the positive predictive value of each method were calculated and used for comparison.
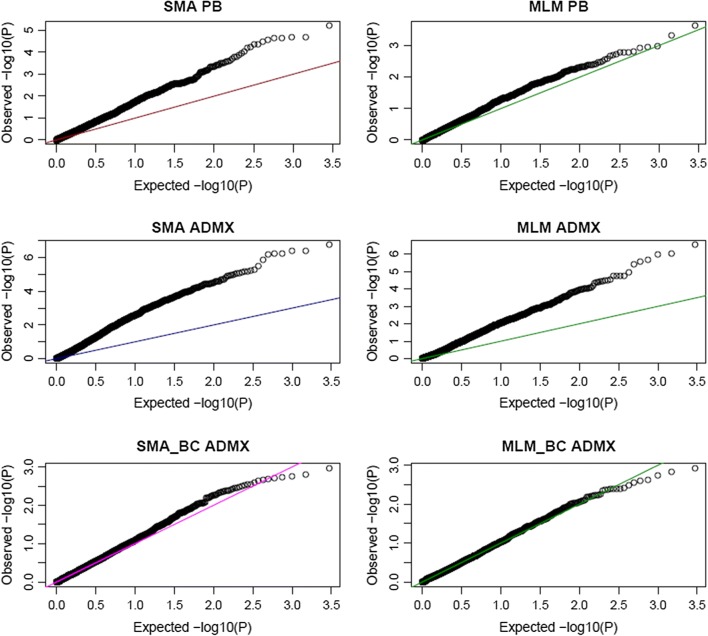
SMR and BMR, both without BC, were ranked as the worst and the best performing approaches, respectively. Our results showed that, while explicit modeling of genetic relationships and BC is essential for models SMR and MLM, BMR can disregard them and yet result in a higher power without compromising its false-positive rate.
This study showed that the Bayesian multiple-regression analysis is robust to population structure and to relationships among study subjects and performs better than a single marker mixed linear model approach.
人群分层和隐性关系是基于人群的关联研究中出现过多假阳性和假阴性的主要原因。已经开发了许多方法来模拟这些混杂因素,并最大程度地减少它们对全基因组关联研究结果的影响。在大多数这些方法中,应用了两阶段方法,其中:(1)方法用于确定样本数据集是否存在群体结构;(2)通过对其进行建模或在每个子群体中单独进行分析来纠正群体结构的影响。本研究的目的是使用贝叶斯多元回归方法评估群体结构对全基因组关联研究准确性和功效的影响。
我们在随机模拟的混合人群中进行了全基因组关联研究。基因组由六个染色体组成,每个染色体有 1000 个标记。15 个分离的数量性状基因座对具有 0.30 遗传力的数量性状的遗传变异做出贡献。评估了三种分析方法(单标记简单回归(SMR),单标记混合线性模型(MLM)和贝叶斯多元回归分析(BMR))中遗传关系和品种组成(BC)的影响。每种方法均在有无 BC 的情况下进行拟合。计算并比较了每种方法的准确性、功效、假阳性率和阳性预测值。
未考虑 BC 的 SMR 和 BMR 分别被评为表现最差和最佳的方法。我们的结果表明,虽然遗传关系和 BC 的显式建模对于 SMR 和 MLM 模型至关重要,但 BMR 可以忽略它们,而在不影响其假阳性率的情况下仍能获得更高的功效。
这项研究表明,贝叶斯多元回归分析对群体结构以及研究对象之间的关系具有鲁棒性,并且比单标记混合线性模型方法表现更好。