Gregor Mendel Institute (GMI), Austrian Academy of Sciences, Vienna, Austria.
Nat Genet. 2012 Jun 17;44(7):825-30. doi: 10.1038/ng.2314.
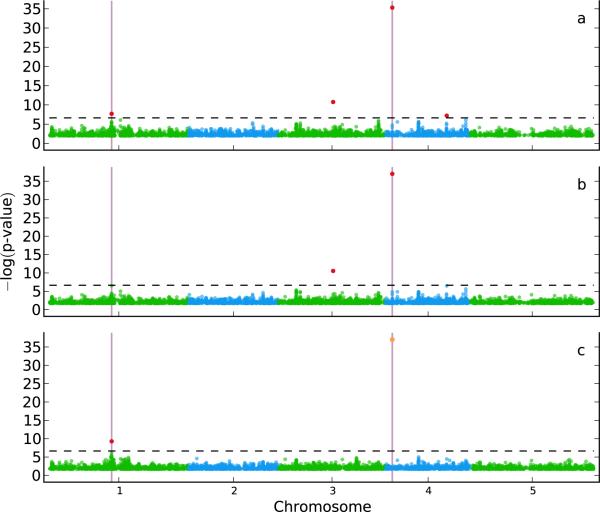
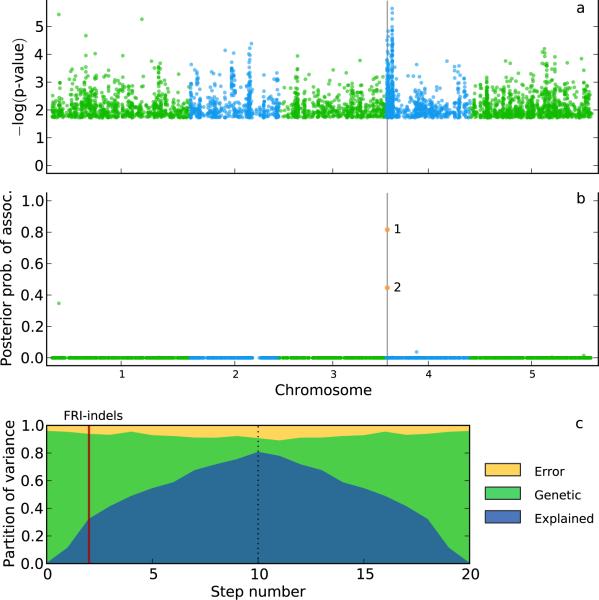
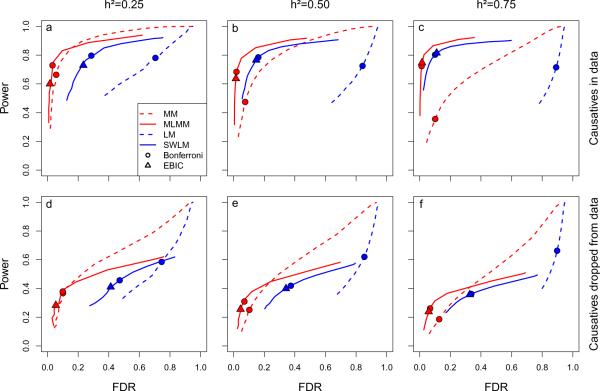
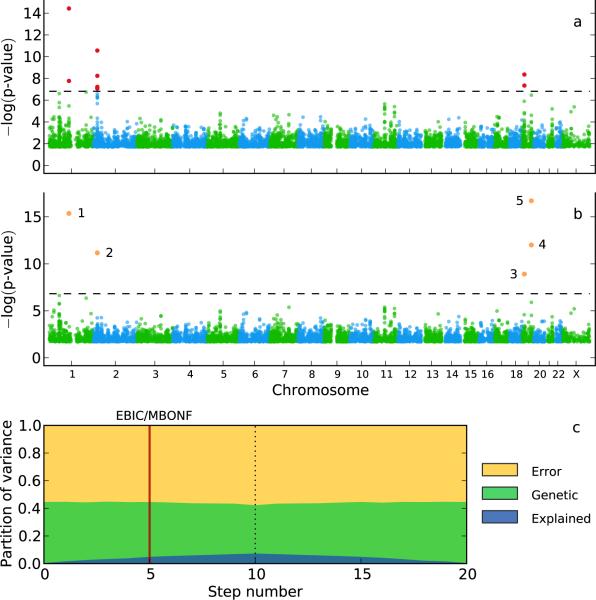
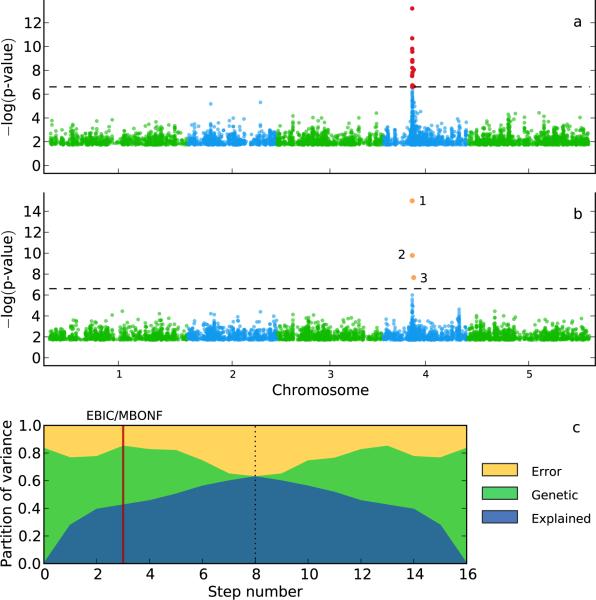
Population structure causes genome-wide linkage disequilibrium between unlinked loci, leading to statistical confounding in genome-wide association studies. Mixed models have been shown to handle the confounding effects of a diffuse background of large numbers of loci of small effect well, but they do not always account for loci of larger effect. Here we propose a multi-locus mixed model as a general method for mapping complex traits in structured populations. Simulations suggest that our method outperforms existing methods in terms of power as well as false discovery rate. We apply our method to human and Arabidopsis thaliana data, identifying new associations and evidence for allelic heterogeneity. We also show how a priori knowledge from an A. thaliana linkage mapping study can be integrated into our method using a Bayesian approach. Our implementation is computationally efficient, making the analysis of large data sets (n > 10,000) practicable.
人口结构导致非连锁基因座间的全基因组连锁不平衡,从而导致全基因组关联研究中的统计混杂。混合模型已被证明能够很好地处理大量小效应基因座弥漫背景的混杂效应,但它们并不总是能解释大效应基因座的情况。在这里,我们提出了一种多基因座混合模型,作为一种在结构群体中定位复杂性状的通用方法。模拟表明,我们的方法在功效和假发现率方面都优于现有方法。我们将该方法应用于人类和拟南芥的数据,鉴定出新的关联和等位基因异质性的证据。我们还展示了如何使用贝叶斯方法将来自拟南芥连锁图谱研究的先验知识整合到我们的方法中。我们的实现方法计算效率高,使对大型数据集(n > 10000)的分析成为可能。