Bioinformatics and Computational Biology Program, Iowa State University, Ames, Iowa, United States of America.
Roy J. Carver Department of Biochemistry, Biophysics and Molecular Biology, Iowa State University, Ames, Iowa, United States of America.
PLoS One. 2018 Jun 20;13(6):e0199225. doi: 10.1371/journal.pone.0199225. eCollection 2018.
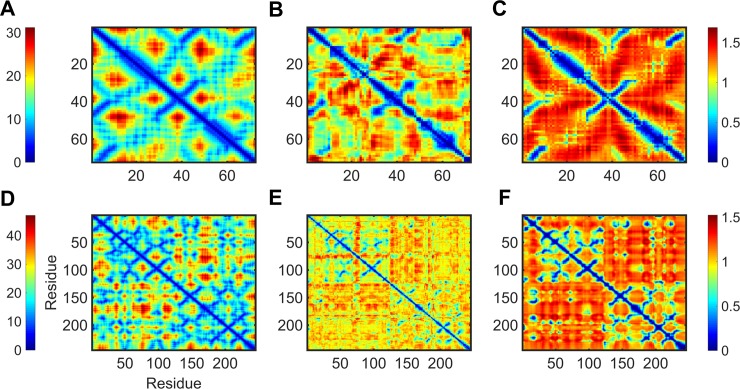
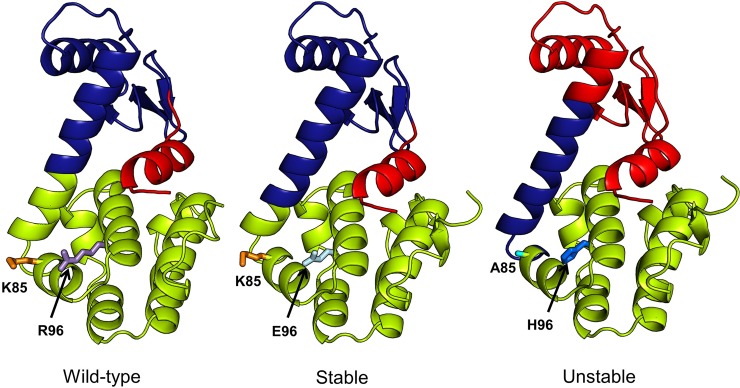
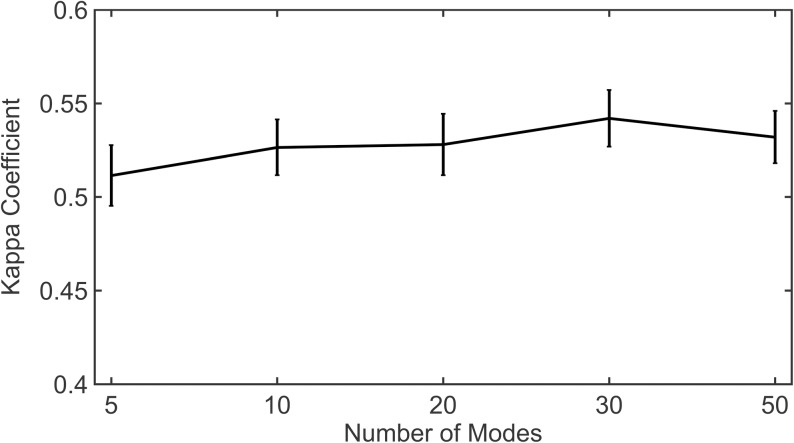
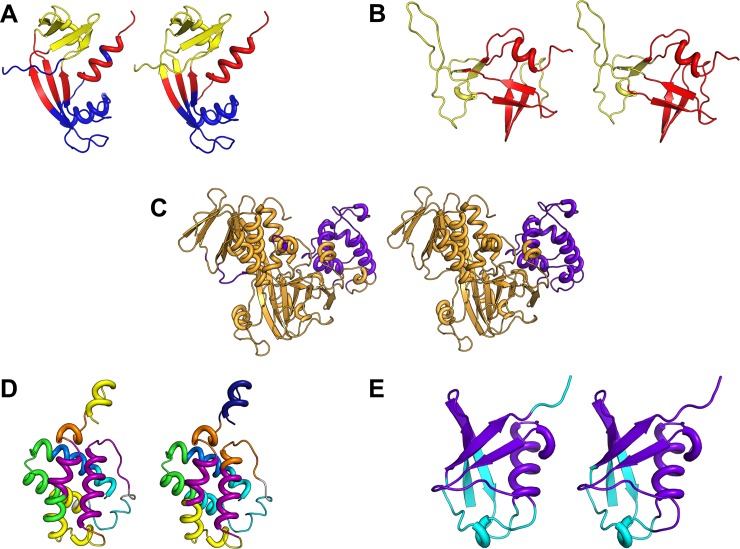
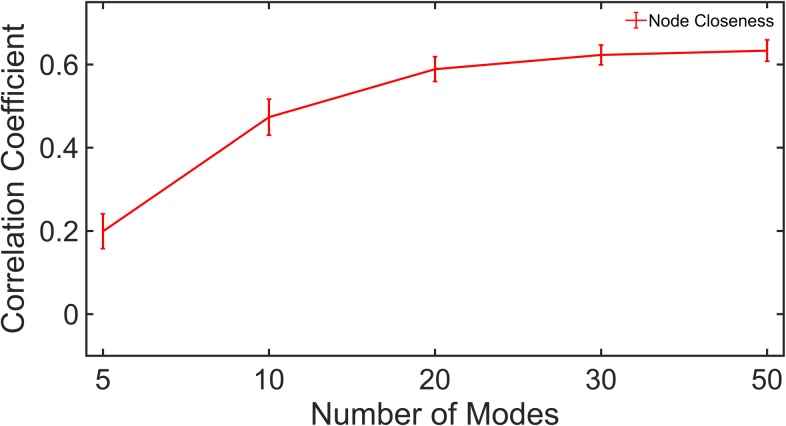
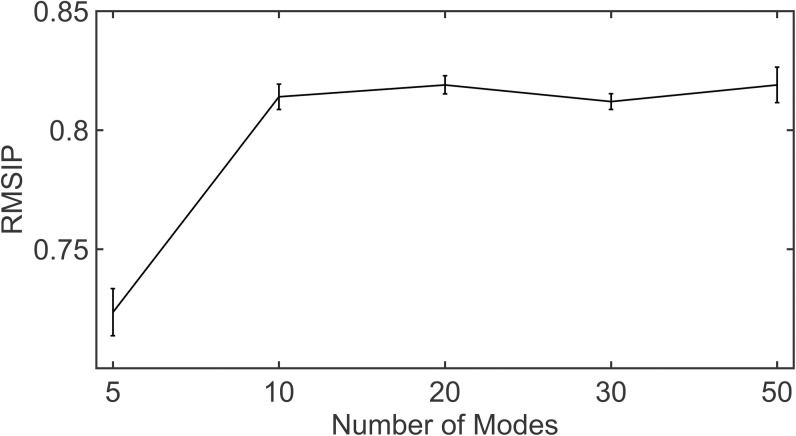
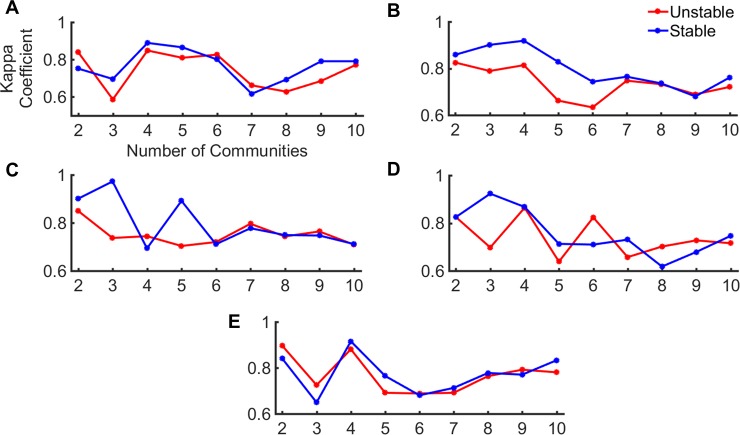
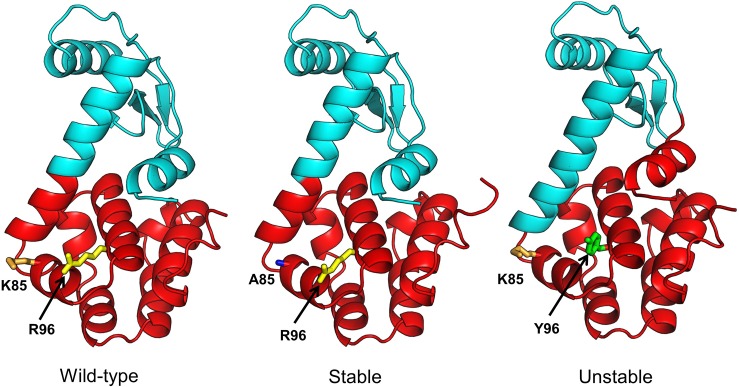
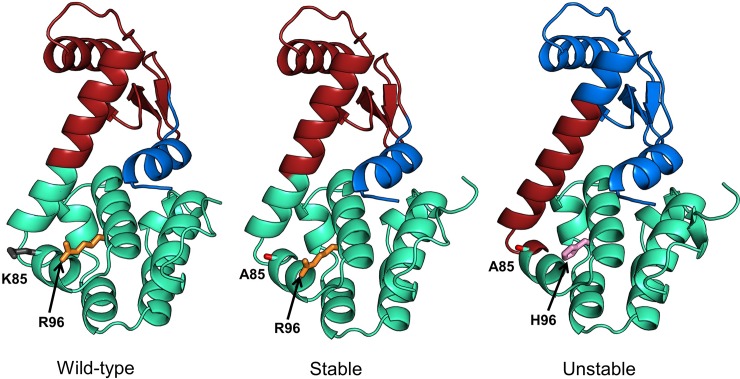
Dynamic communities in proteins comprise the cohesive structural units that individually exhibit rigid body motions. These can correspond to structural domains, but are usually smaller parts that move with respect to one another in a protein's internal motions, key to its functional dynamics. Previous studies emphasized their importance to understand the nature of ligand-induced allosteric regulation. These studies reported that mutations to key community residues can hinder transmission of allosteric signals among the communities. Usually molecular dynamic (MD) simulations (~ 100 ns or longer) have been used to identify the communities-a demanding task for larger proteins. In the present study, we propose that dynamic communities obtained from MD simulations can also be obtained alternatively with simpler models-the elastic network models (ENMs). To verify this premise, we compare the specific communities obtained from MD and ENMs for 44 proteins. We evaluate the correspondence in communities from the two methods and compute the extent of agreement in the dynamic cross-correlation data used for community detection. Our study reveals a strong correspondence between the communities from MD and ENM and also good agreement for the residue cross-correlations. Importantly, we observe that the dynamic communities from MD can be closely reproduced with ENMs. With ENMs, we also compare the community structures of stable and unstable mutant forms of T4 Lysozyme with its wild-type. We find that communities for unstable mutants show substantially poorer agreement with the wild-type communities than do stable mutants, suggesting such ENM-based community structures can serve as a means to rapidly identify deleterious mutants.
蛋白质中的动态社区由具有刚体运动的凝聚结构单元组成。这些可以对应结构域,但通常是较小的部分,它们在蛋白质的内部运动中相对于彼此移动,这是其功能动态的关键。先前的研究强调了它们对于理解配体诱导的变构调节的性质的重要性。这些研究报告说,关键社区残基的突变会阻碍社区之间的变构信号传递。通常使用分子动力学 (MD) 模拟(~100ns 或更长时间)来识别社区-对于较大的蛋白质来说这是一项艰巨的任务。在本研究中,我们提出可以使用更简单的模型(弹性网络模型 (ENM))替代 MD 模拟来获得动态社区。为了验证这一前提,我们将 MD 和 ENM 获得的特定社区进行了比较,涉及 44 种蛋白质。我们从两种方法中评估社区的对应性,并计算用于社区检测的动态互相关数据的一致性程度。我们的研究揭示了 MD 和 ENM 中的社区之间存在很强的对应关系,并且在残基互相关方面也具有很好的一致性。重要的是,我们观察到 MD 中的动态社区可以用 ENM 紧密再现。通过 ENM,我们还比较了 T4 溶菌酶及其野生型的稳定和不稳定突变体的社区结构。我们发现,不稳定突变体的社区与野生型社区的一致性要差得多,这表明这种基于 ENM 的社区结构可以作为快速识别有害突变体的一种手段。