Business Intelligence and Machine Learning Research Group-GPIN, School of Technology, PUCRS, Av. Ipiranga, 6681, Building 32, Room 628, Porto Alegre, RS, Brazil.
Bioinformatics and Biossystems Modeling and Simulation Lab-LABIO, School of Technology, PUCRS, Av. Ipiranga, 6681, Building 32, Room 602, Porto Alegre, RS, Brazil.
BMC Bioinformatics. 2018 Jun 22;19(1):235. doi: 10.1186/s12859-018-2222-2.
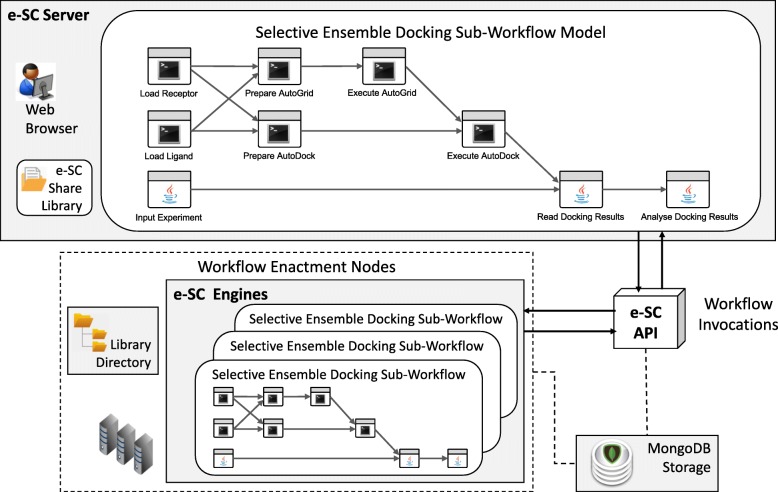
In the rational drug design process, an ensemble of conformations obtained from a molecular dynamics simulation plays a crucial role in docking experiments. Some studies have found that Fully-Flexible Receptor (FFR) models predict realistic binding energy accurately and improve scoring to enhance selectiveness. At the same time, methods have been proposed to reduce the high computational costs involved in considering the explicit flexibility of proteins in receptor-ligand docking. This study introduces a novel method to optimize ensemble docking-based experiments by reducing the size of an InhA FFR model at docking runtime and scaling docking workflow invocations on cloud virtual machines.
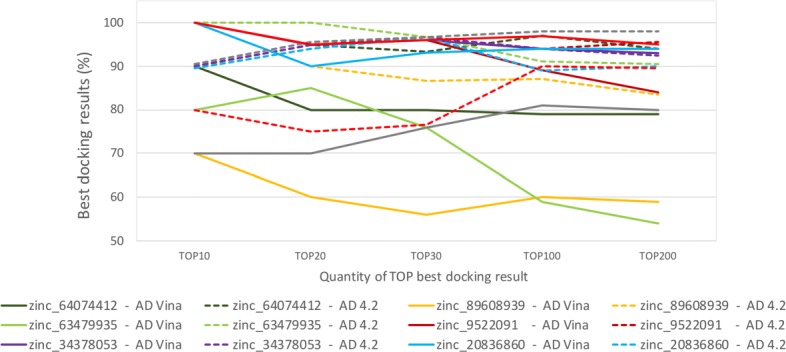
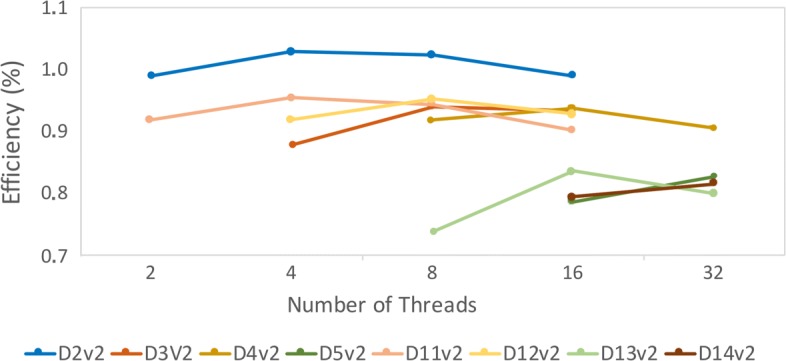
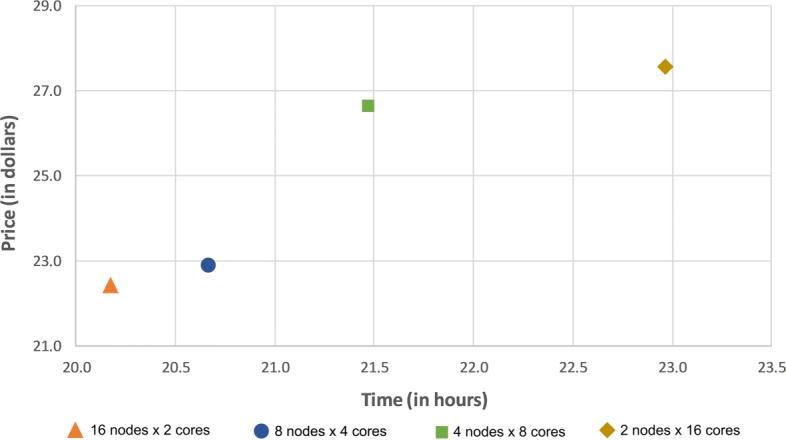
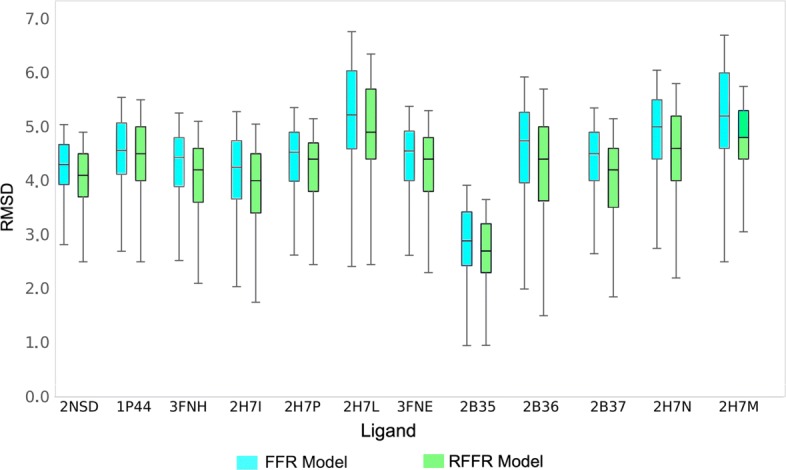
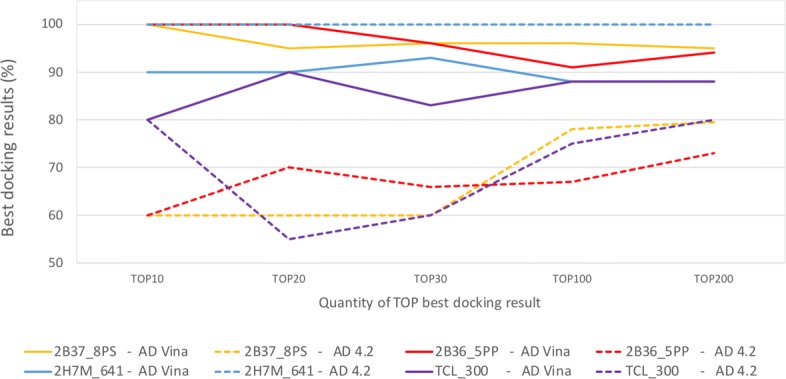
First, in order to find the most affordable cost-benefit pool of virtual machines, we evaluated the performance of the docking workflow invocations in different configurations of Azure instances. Second, we validated the gains obtained by the proposed method based on the quality of the Reduced Fully-Flexible Receptor (RFFR) models produced using AutoDock4.2. The analyses show that the proposed method reduced the model size by approximately 50% while covering at least 86% of the best docking results from the 74 ligands tested. Third, we tested our novel method using AutoDock Vina, a different docking software, and showed the positive accuracy achieved in the resulting RFFR models. Finally, our results demonstrated that the method proposed optimized ensemble docking experiments and is applicable to different docking software. In addition, it detected new binding modes, which would be unreachable if employing only the rigid structure used to generate the InhA FFR model.
Our results showed that the selective method is a valuable strategy for optimizing ensemble docking-based experiments using different docking software. The RFFR models produced by discarding non-promising snapshots from the original model are accurately shaped for a larger number of ligands, and the elapsed time spent in the ensemble docking experiments are considerably reduced.
在合理药物设计过程中,分子动力学模拟得到的构象集合在对接实验中起着至关重要的作用。一些研究发现,完全柔性受体(FFR)模型可以准确地预测实际的结合能,并通过改进评分来提高选择性。同时,也提出了一些方法来降低在受体-配体对接中考虑蛋白质显式灵活性所涉及的高计算成本。本研究通过在对接运行时缩小 InhA FFR 模型的大小,并在云虚拟机上扩展对接工作流程的调用,介绍了一种优化基于集合的对接实验的新方法。
首先,为了找到最具成本效益的虚拟机池,我们评估了对接工作流程在不同 Azure 实例配置下的性能。其次,我们根据使用 AutoDock4.2 生成的简化完全柔性受体(RFFR)模型的质量,验证了所提出方法的收益。分析表明,该方法将模型尺寸缩小了约 50%,同时覆盖了测试的 74 个配体中至少 86%的最佳对接结果。第三,我们使用不同的对接软件 AutoDock Vina 测试了我们的新方法,并展示了在所得 RFFR 模型中实现的正准确性。最后,我们的结果表明,该方法优化了基于集合的对接实验,适用于不同的对接软件。此外,它还检测到了新的结合模式,如果只使用生成 InhA FFR 模型的刚性结构,这些模式将无法触及。
我们的结果表明,选择性方法是一种使用不同对接软件优化基于集合的对接实验的有价值策略。从原始模型中丢弃非有希望的快照生成的 RFFR 模型可以准确地适应更多的配体,并且在集合对接实验中花费的时间大大减少。