Department of Biochemistry, University of Cambridge, Cambridge CB2 1GA, UK.
Department of Drugs and Medicines; School of Pharmacy; Federal University of Rio de Janeiro, Rio de Janeiro 21949-900, RJ, Brazil.
Int J Mol Sci. 2019 Sep 15;20(18):4574. doi: 10.3390/ijms20184574.
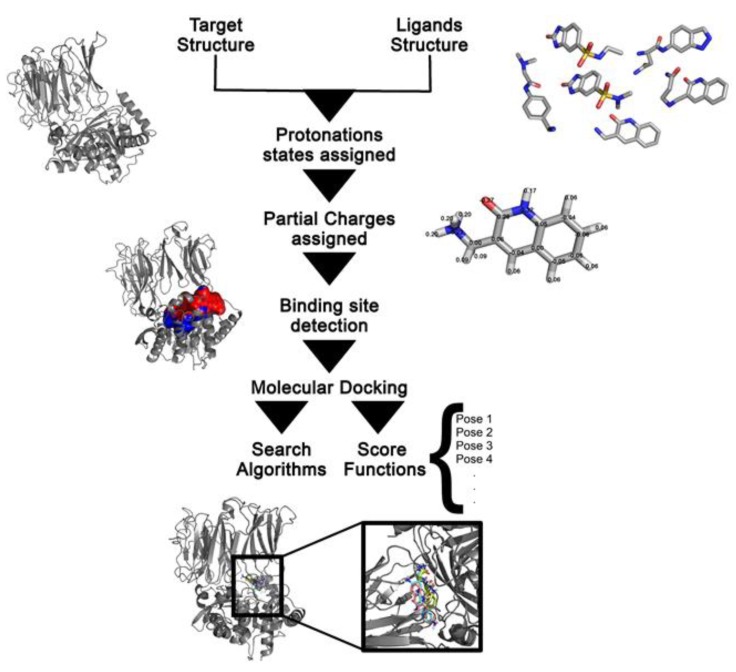
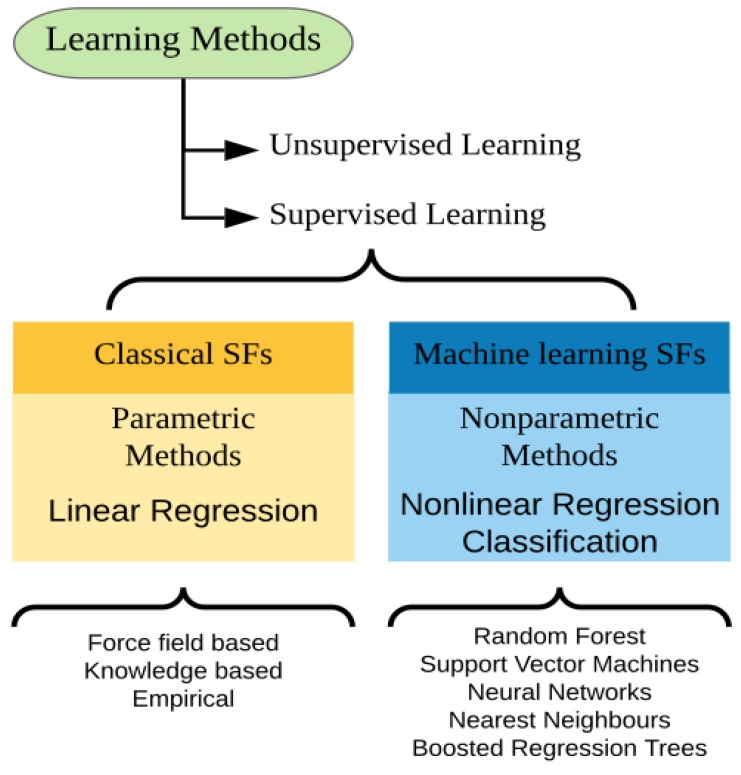
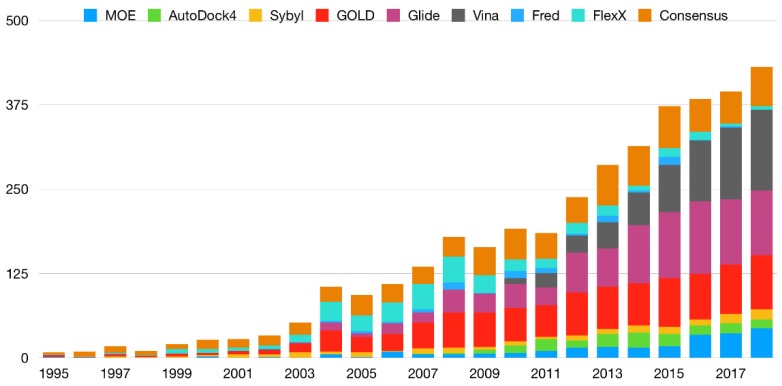
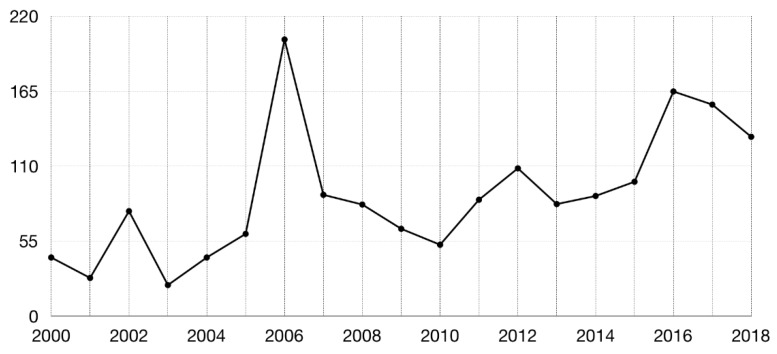
Molecular docking has been widely employed as a fast and inexpensive technique in the past decades, both in academic and industrial settings. Although this discipline has now had enough time to consolidate, many aspects remain challenging and there is still not a straightforward and accurate route to readily pinpoint true ligands among a set of molecules, nor to identify with precision the correct ligand conformation within the binding pocket of a given target molecule. Nevertheless, new approaches continue to be developed and the volume of published works grows at a rapid pace. In this review, we present an overview of the method and attempt to summarise recent developments regarding four main aspects of molecular docking approaches: (i) the available benchmarking sets, highlighting their advantages and caveats, (ii) the advances in consensus methods, (iii) recent algorithms and applications using fragment-based approaches, and (iv) the use of machine learning algorithms in molecular docking. These recent developments incrementally contribute to an increase in accuracy and are expected, given time, and together with advances in computing power and hardware capability, to eventually accomplish the full potential of this area.
分子对接在过去几十年中被广泛应用于学术和工业领域,作为一种快速且廉价的技术。尽管该学科现在已经有足够的时间来巩固,但仍有许多方面具有挑战性,并且仍然没有一种直接而准确的方法来确定一组分子中的真正配体,也无法精确识别给定靶分子结合口袋中正确的配体构象。然而,新的方法仍在不断发展,发表的作品数量也在迅速增长。在这篇综述中,我们介绍了该方法,并尝试总结了分子对接方法的四个主要方面的最新进展:(i)现有的基准测试集,突出其优点和缺点,(ii)共识方法的进展,(iii)基于片段的方法的最新算法和应用,以及(iv)机器学习算法在分子对接中的应用。这些最新的进展逐步提高了准确性,并有望随着时间的推移,以及计算能力和硬件能力的进步,最终实现该领域的全部潜力。