Riesco Raúl, Carro Lorena, Román-Ponce Brenda, Prieto Carlos, Blom Jochen, Klenk Hans-Peter, Normand Philippe, Trujillo Martha E
Departament of Microbiology and Genetics, Edificio Departamental, University of Salamanca, Salamanca, Spain.
Servicio de Bioinformática, NUCLEUS, Edificio I+D+i, University of Salamanca, Salamanca, Spain.
Front Microbiol. 2018 Jun 25;9:1360. doi: 10.3389/fmicb.2018.01360. eCollection 2018.
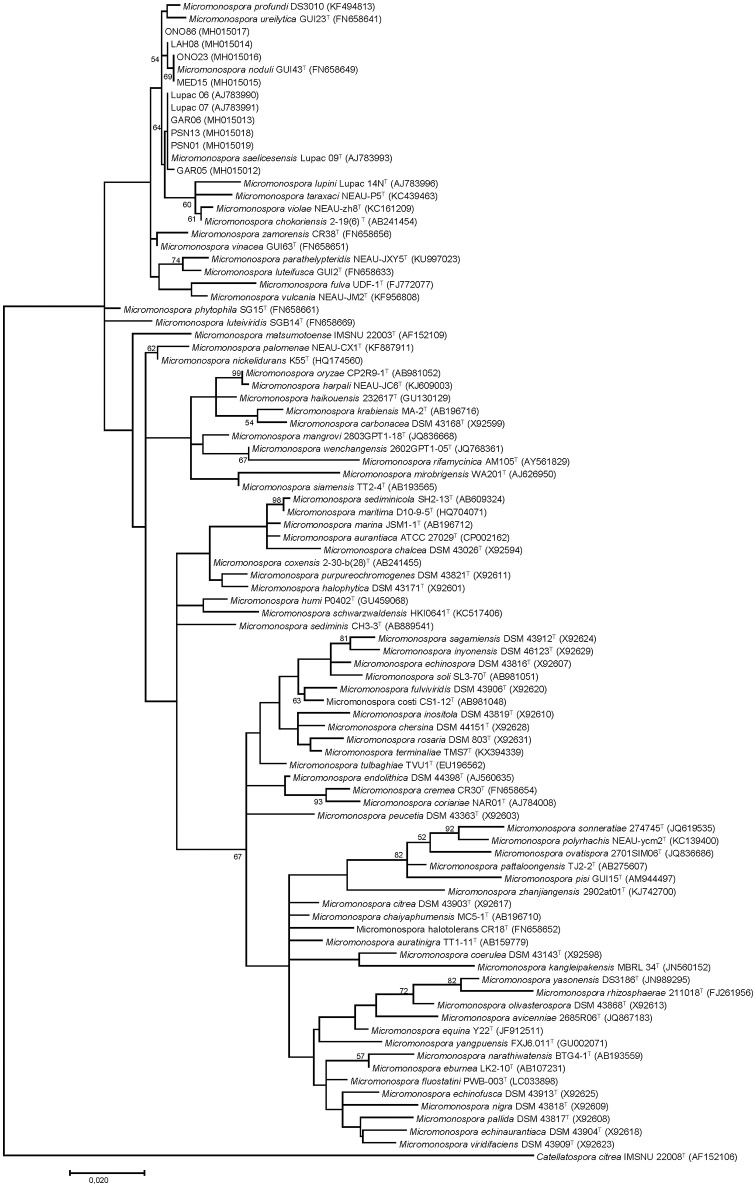
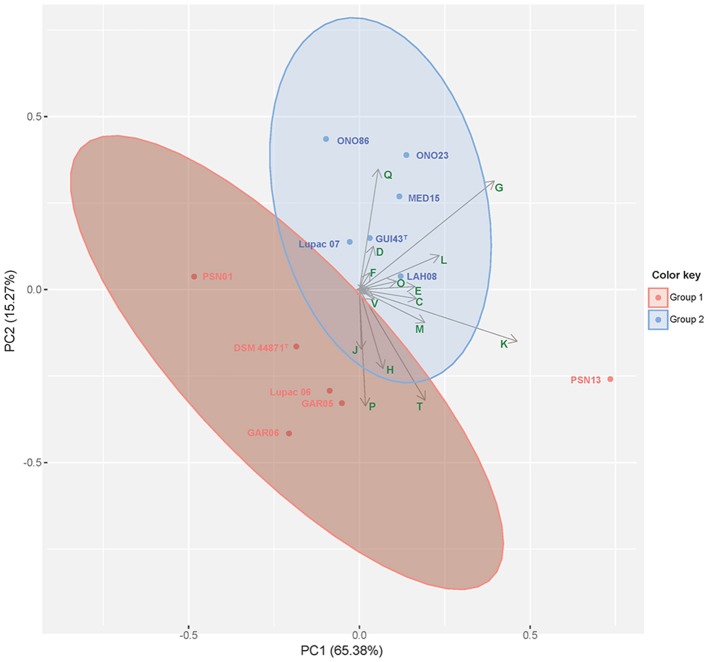
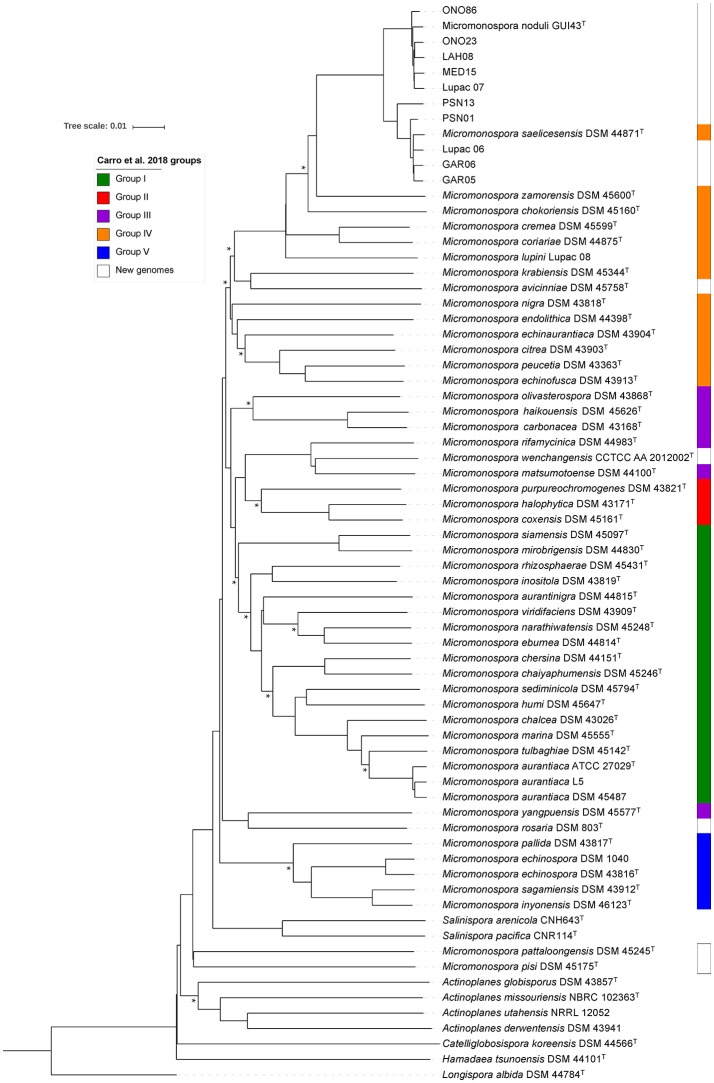
The type isolates of species and are Gram-stain positive actinobacteria that were originally isolated from nitrogen fixing nodules of the legumes and , respectively. These two species are very closely related and questions arise as to whether they should be merged into a single species. To better delineate the relationship of and , 10 strains isolated from plant tissue (nodules and leaves) and identified by their 16S rRNA gene sequences as either or , based on a cut-off value of ≥99.5% were selected for whole-genome sequencing and compared with the type strains of Lupac 09 and GUI43 using overall genome relatedness indices (OGRI) which included ANI, OrthoANI and digital DNA-DNA hybridization. Whole- and core-genome phylogenomic analyses were also carried out. These results were compared with the topologies of the 16S rRNA and gene phylogenies. Good correlation was found between all trees except for the 16S rRNA gene. Overall results also supported the current classification of and as separate species. Especially useful was the core-genome phylogenetic analyses based on 92 genes and the dDDH results which were highly correlated. The importance of using more than one strain for a better definition of a species was also shown. A series of phenotypic assays performed at different times were compared with predictions based on genomic data. phenotypic tests showed discrepancies among the independent studies, confirming the lack of reproducibility even when tests were performed in the same laboratory. On the other hand, the use of predictions proved useful for defining a stable phenotype profile among the strains analyzed. These results provide a working framework for defining species at the genomic and phenotypic level.
物种 和 的模式菌株是革兰氏染色阳性放线菌,最初分别从豆科植物 和 的固氮根瘤中分离得到。这两个物种关系非常密切,因此出现了它们是否应合并为一个单一物种的问题。为了更好地描述 和 的关系,从植物组织(根瘤和叶片)中分离出10株菌株,并根据16S rRNA基因序列将其鉴定为 或 ,基于≥99.5%的截止值,选择这些菌株进行全基因组测序,并使用包括ANI、OrthoANI和数字DNA-DNA杂交在内的全基因组相关性指数(OGRI)与 Lupac 09和 GUI43的模式菌株进行比较。还进行了全基因组和核心基因组系统发育分析。将这些结果与16S rRNA和 基因系统发育树的拓扑结构进行比较。除16S rRNA基因外,在所有树之间都发现了良好的相关性。总体结果也支持目前将 和 分类为不同物种的分类方法。基于92个基因的核心基因组系统发育分析和高度相关的dDDH结果特别有用。还显示了使用多个菌株以更好地定义一个物种的重要性。将在不同时间进行的一系列 表型试验与基于基因组数据的 预测进行了比较。 表型试验在独立研究之间显示出差异,证实了即使在同一实验室进行试验也缺乏可重复性。另一方面, 使用预测被证明有助于在分析的菌株中定义稳定的表型特征。这些结果为在基因组和表型水平上定义 物种提供了一个工作框架。