Bodnar Colleen N, Morganti Josh M, Bachstetter Adam D
Spinal Cord & Brain Injury Research Center, University of Kentucky; Department of Neuroscience, University of Kentucky, Lexington, KY, USA.
Department of Neuroscience, University of Kentucky; Sanders-Brown Center on Aging, University of Kentucky, Lexington, KY, USA.
Neural Regen Res. 2018 Oct;13(10):1693-1704. doi: 10.4103/1673-5374.238604.
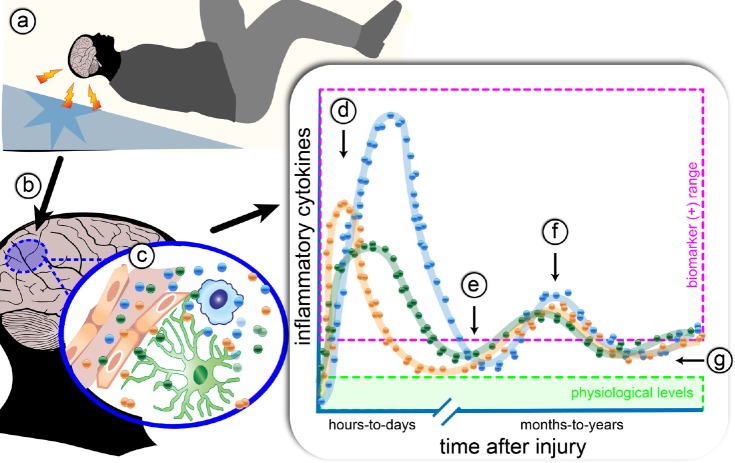
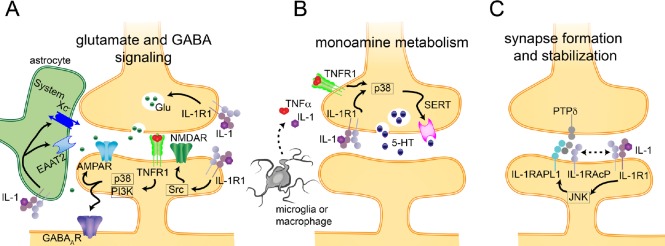
A substantial number of individuals have long-lasting adverse effects from a traumatic brain injury (TBI). Depression is one of these long-term complications that influences many aspects of life. Depression can limit the ability to return to work, and even worsen cognitive function and contribute to dementia. The mechanistic cause for the increased depression risk associated with a TBI remains to be defined. As TBI results in chronic neuroinflammation, and priming of glia to a secondary challenge, the inflammatory theory of depression provides a promising framework for investigating the cause of depression following a TBI. Increases in cytokines similar to those seen in depression in the general population are also increased following a TBI. Biomarker levels of cytokines peak within hours-to-days after the injury, yet pro-inflammatory cytokines may still be elevated above physiological levels months-to-years following TBI, which is the time frame in which post-TBI depression can persist. As tumor necrosis factor α and interleukin 1 can signal directly at the neuronal synapse, pathophysiological levels of these cytokines can detrimentally alter neuronal synaptic physiology. The purpose of this review is to outline the current evidence for the inflammatory hypothesis of depression specifically as it relates to depression following a TBI. Moreover, we will illustrate the potential synaptic mechanisms by which tumor necrosis factor α and interleukin 1 could contribute to depression. The association of inflammation with the development of depression is compelling; however, in the context of post-TBI depression, the role of inflammation is understudied. This review attempts to highlight the need to understand and treat the psychological complications of a TBI, potentially by neuroimmune modulation, as the neuropsychiatric disabilities can have a great impact on the rehabilitation from the injury, and overall quality of life.
相当一部分人因创伤性脑损伤(TBI)而产生长期不良影响。抑郁症是这些长期并发症之一,会影响生活的许多方面。抑郁症会限制重返工作岗位的能力,甚至会使认知功能恶化并导致痴呆。与TBI相关的抑郁症风险增加的机制原因仍有待确定。由于TBI会导致慢性神经炎症,并使胶质细胞对二次挑战产生预激,抑郁症的炎症理论为研究TBI后抑郁症的病因提供了一个有前景的框架。与普通人群中抑郁症患者所见相似的细胞因子水平在TBI后也会升高。细胞因子的生物标志物水平在损伤后数小时至数天内达到峰值,但促炎细胞因子在TBI后的数月至数年仍可能高于生理水平,这正是TBI后抑郁症可能持续的时间范围。由于肿瘤坏死因子α和白细胞介素1可直接在神经元突触处发出信号,这些细胞因子的病理生理水平会有害地改变神经元突触生理。本综述的目的是概述抑郁症炎症假说的当前证据,特别是与TBI后抑郁症相关的证据。此外,我们将阐述肿瘤坏死因子α和白细胞介素1可能导致抑郁症的潜在突触机制。炎症与抑郁症发生之间的关联很有说服力;然而,在TBI后抑郁症的背景下,炎症的作用尚未得到充分研究。本综述试图强调理解和治疗TBI心理并发症的必要性,可能通过神经免疫调节来实现,因为神经精神残疾会对损伤后的康复和整体生活质量产生重大影响。