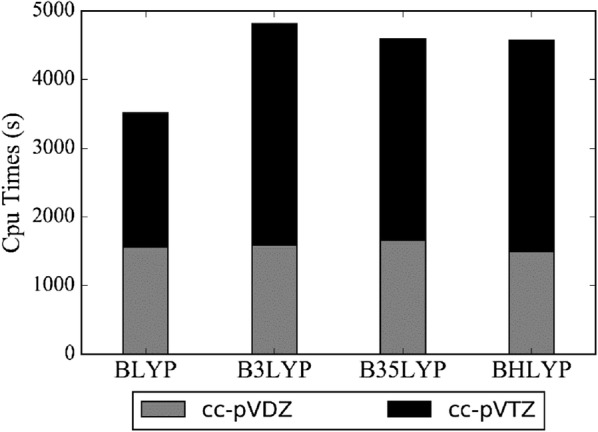
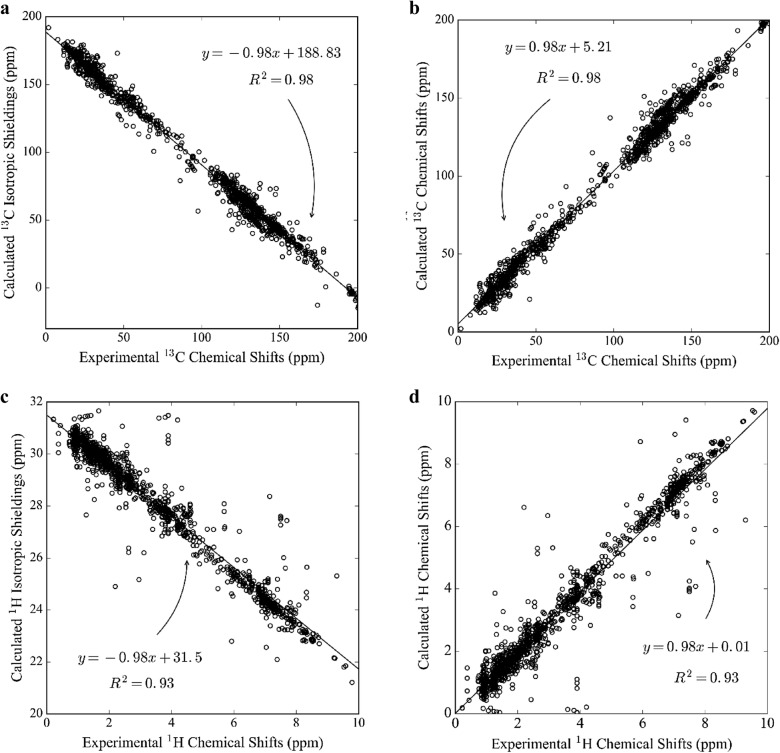
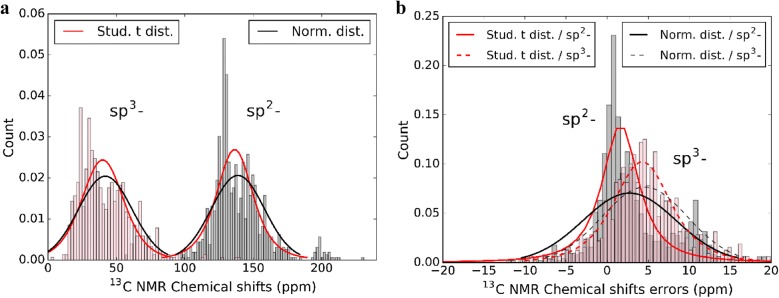
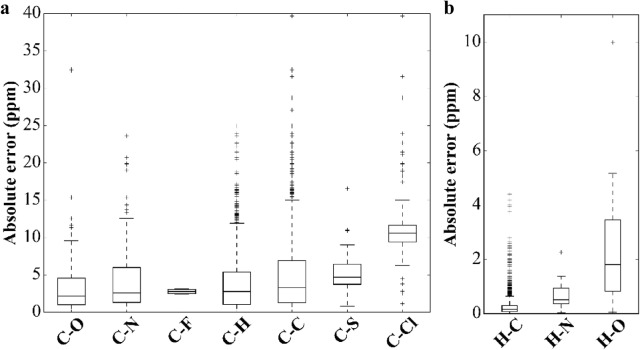
Yesiltepe Yasemin, Nuñez Jamie R, Colby Sean M, Thomas Dennis G, Borkum Mark I, Reardon Patrick N, Washton Nancy M, Metz Thomas O, Teeguarden Justin G, Govind Niranjan, Renslow Ryan S
The Gene and Linda Voiland School of Chemical Engineering and Bioengineering, Washington State University, Pullman, WA, USA.
Earth and Biological Sciences Division, Pacific Northwest National Laboratory, Richland, WA, USA.
J Cheminform. 2018 Oct 26;10(1):52. doi: 10.1186/s13321-018-0305-8.
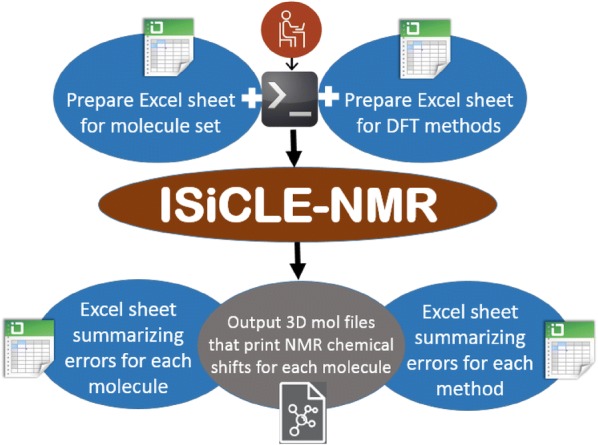
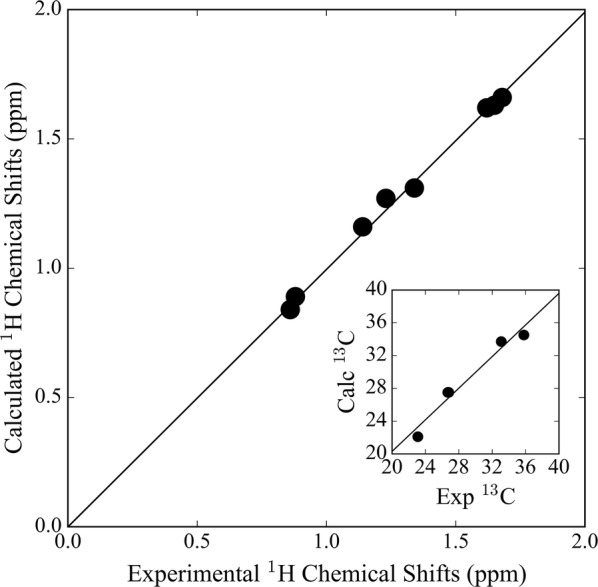
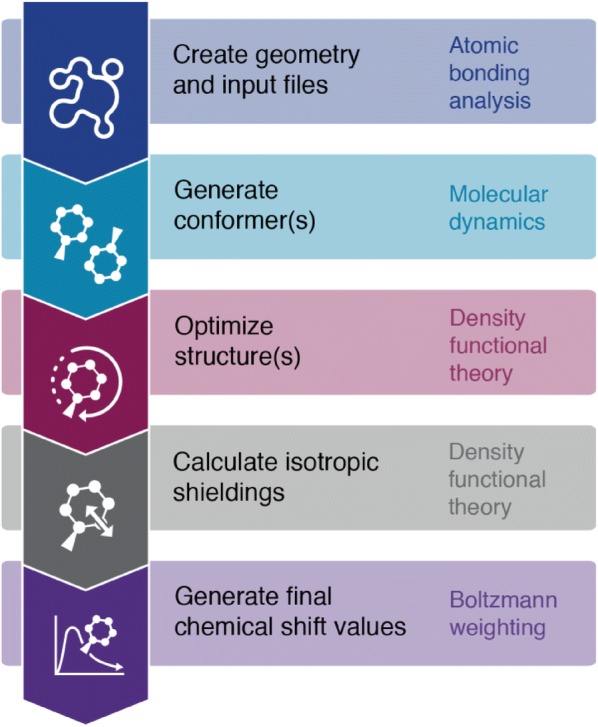
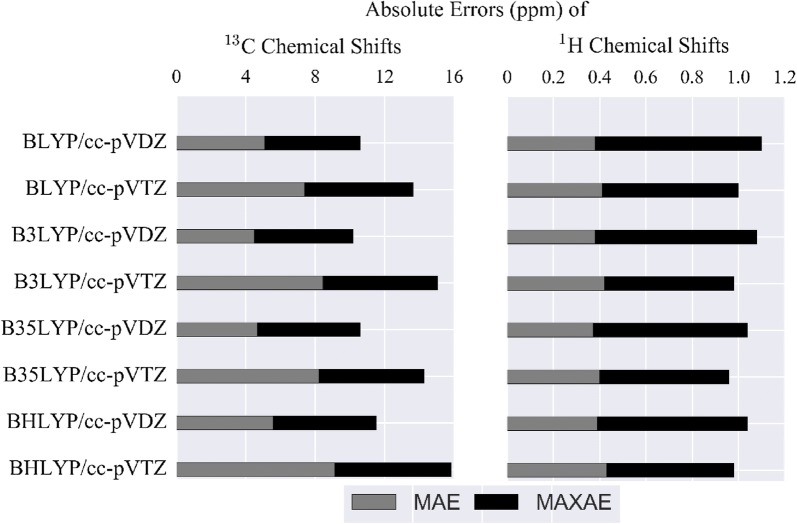
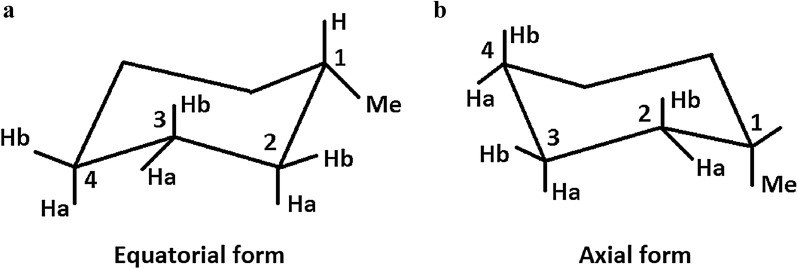
When using nuclear magnetic resonance (NMR) to assist in chemical identification in complex samples, researchers commonly rely on databases for chemical shift spectra. However, authentic standards are typically depended upon to build libraries experimentally. Considering complex biological samples, such as blood and soil, the entirety of NMR spectra required for all possible compounds would be infeasible to ascertain due to limitations of available standards and experimental processing time. As an alternative, we introduce the in silico Chemical Library Engine (ISiCLE) NMR chemical shift module to accurately and automatically calculate NMR chemical shifts of small organic molecules through use of quantum chemical calculations. ISiCLE performs density functional theory (DFT)-based calculations for predicting chemical properties-specifically NMR chemical shifts in this manuscript-via the open source, high-performance computational chemistry software, NWChem. ISiCLE calculates the NMR chemical shifts of sets of molecules using any available combination of DFT method, solvent, and NMR-active nuclei, using both user-selected reference compounds and/or linear regression methods. Calculated NMR chemical shifts are provided to the user for each molecule, along with comparisons with respect to a number of metrics commonly used in the literature. Here, we demonstrate ISiCLE using a set of 312 molecules, ranging in size up to 90 carbon atoms. For each, calculation of NMR chemical shifts have been performed with 8 different levels of DFT theory, and with solvation effects using the implicit solvent Conductor-like Screening Model. The DFT method dependence of the calculated chemical shifts have been systematically investigated through benchmarking and subsequently compared to experimental data available in the literature. Furthermore, ISiCLE has been applied to a set of 80 methylcyclohexane conformers, combined via Boltzmann weighting and compared to experimental values. We demonstrate that our protocol shows promise in the automation of chemical shift calculations and, ultimately, the expansion of chemical shift libraries.
在使用核磁共振(NMR)辅助复杂样品的化学鉴定时,研究人员通常依赖化学位移光谱数据库。然而,构建库通常需要依靠真实标准品进行实验。考虑到复杂的生物样品,如血液和土壤,由于现有标准品的局限性和实验处理时间的限制,确定所有可能化合物所需的完整NMR光谱是不可行的。作为替代方案,我们引入了计算机化学库引擎(ISiCLE)NMR化学位移模块,通过使用量子化学计算来准确自动地计算小分子有机化合物的NMR化学位移。ISiCLE通过开源的高性能计算化学软件NWChem,进行基于密度泛函理论(DFT)的计算,以预测化学性质,在此手稿中具体为NMR化学位移。ISiCLE使用DFT方法、溶剂和NMR活性核的任何可用组合,通过用户选择的参考化合物和/或线性回归方法,计算分子组的NMR化学位移。为每个分子向用户提供计算得到的NMR化学位移,并与文献中常用的一些指标进行比较。在此,我们使用一组312个分子(大小范围高达90个碳原子)来演示ISiCLE。对于每个分子,已使用8种不同水平的DFT理论进行NMR化学位移计算,并使用类似导体的屏蔽模型隐式溶剂考虑溶剂化效应。通过基准测试系统地研究了计算化学位移对DFT方法的依赖性,并随后与文献中可用的实验数据进行了比较。此外,ISiCLE已应用于一组80个甲基环己烷构象异构体,通过玻尔兹曼加权进行组合并与实验值进行比较。我们证明,我们的方案在化学位移计算自动化以及最终化学位移库扩展方面显示出前景。