Brockherde Felix, Vogt Leslie, Li Li, Tuckerman Mark E, Burke Kieron, Müller Klaus-Robert
Machine Learning Group, Technische Universität Berlin, Marchstraße 23, 10587, Berlin, Germany.
Max-Planck-Institut für Mikrostrukturphysik, Weinberg 2, 06120, Halle, Germany.
Nat Commun. 2017 Oct 11;8(1):872. doi: 10.1038/s41467-017-00839-3.
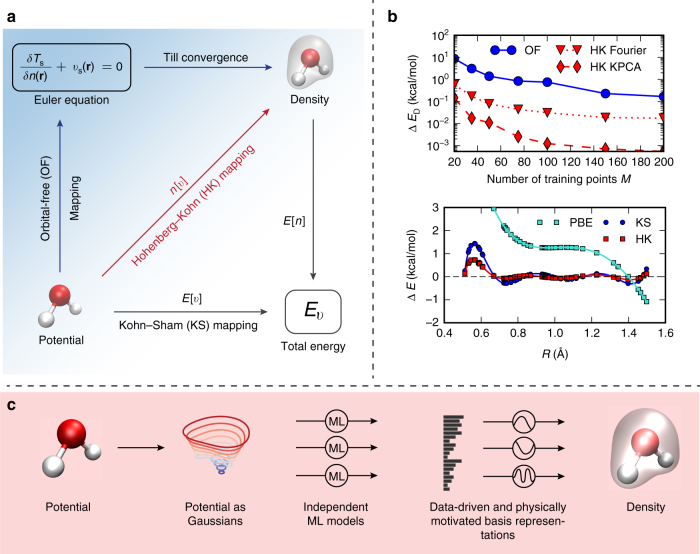
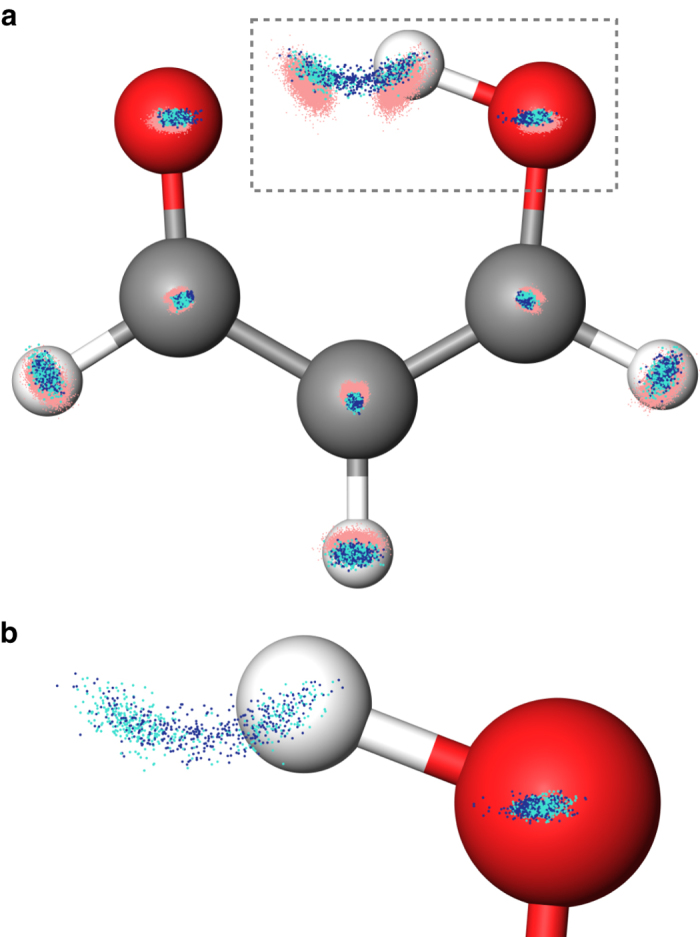
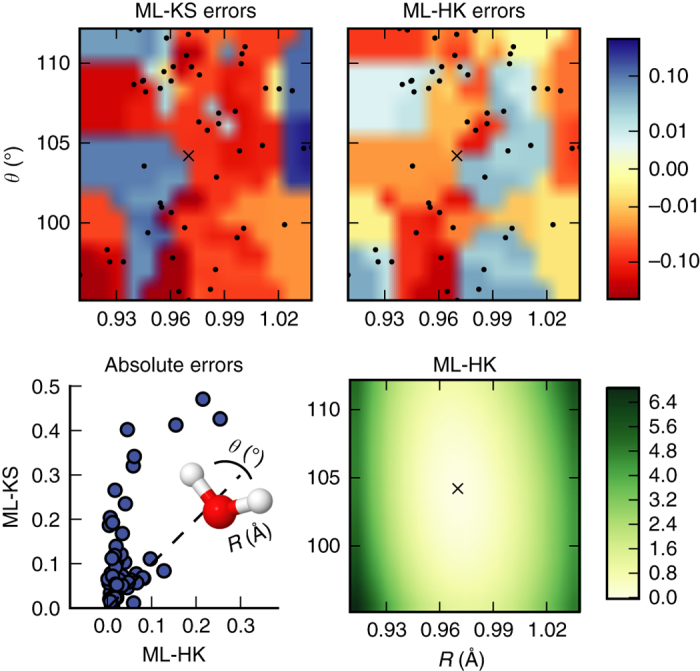
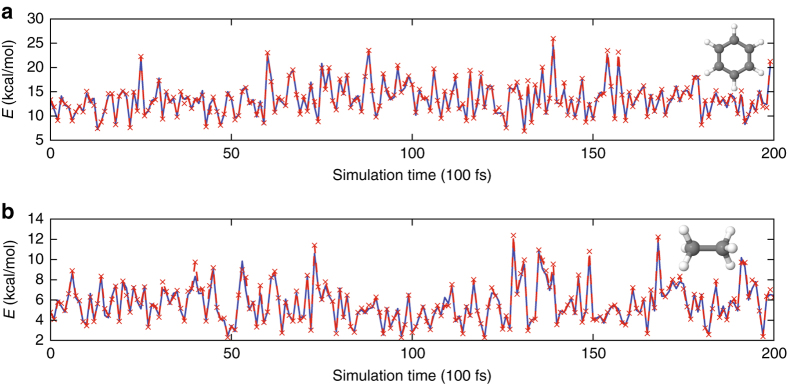
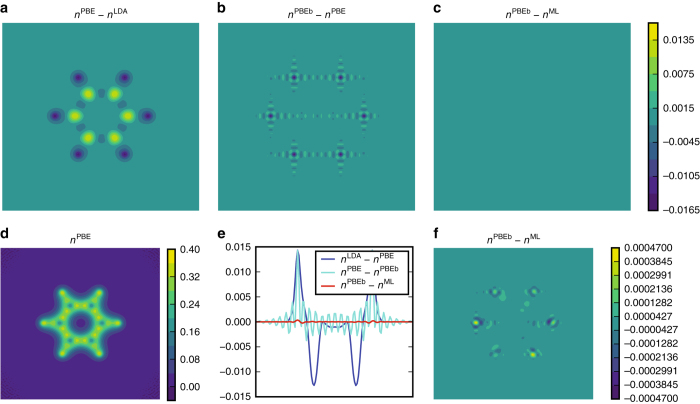
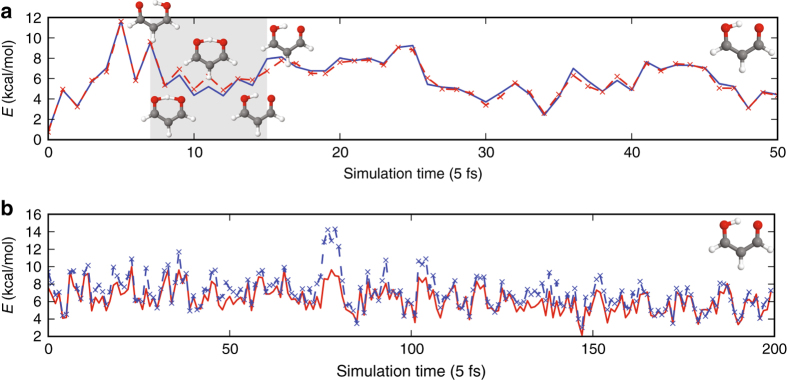
Last year, at least 30,000 scientific papers used the Kohn-Sham scheme of density functional theory to solve electronic structure problems in a wide variety of scientific fields. Machine learning holds the promise of learning the energy functional via examples, bypassing the need to solve the Kohn-Sham equations. This should yield substantial savings in computer time, allowing larger systems and/or longer time-scales to be tackled, but attempts to machine-learn this functional have been limited by the need to find its derivative. The present work overcomes this difficulty by directly learning the density-potential and energy-density maps for test systems and various molecules. We perform the first molecular dynamics simulation with a machine-learned density functional on malonaldehyde and are able to capture the intramolecular proton transfer process. Learning density models now allows the construction of accurate density functionals for realistic molecular systems.Machine learning allows electronic structure calculations to access larger system sizes and, in dynamical simulations, longer time scales. Here, the authors perform such a simulation using a machine-learned density functional that avoids direct solution of the Kohn-Sham equations.
去年,至少3万篇科学论文使用密度泛函理论的科恩-沈(Kohn-Sham)方法来解决各种科学领域中的电子结构问题。机器学习有望通过示例学习能量泛函,从而无需求解科恩-沈方程。这将大幅节省计算机时间,使更大的系统和/或更长的时间尺度得以处理,但机器学习该泛函的尝试一直受到求其导数需求的限制。本研究通过直接学习测试系统和各种分子的密度-势以及能量-密度图克服了这一困难。我们首次使用机器学习的密度泛函对丙二醛进行分子动力学模拟,并能够捕捉分子内质子转移过程。学习密度模型现在允许为实际分子系统构建精确的密度泛函。机器学习使电子结构计算能够处理更大的系统规模,并且在动力学模拟中能够处理更长的时间尺度。在此,作者使用避免直接求解科恩-沈方程的机器学习密度泛函进行了这样的模拟。