Barel Gal, Herwig Ralf
Department Computational Molecular Biology, Max Planck Institute for Molecular Genetics, Berlin, Germany.
Front Genet. 2018 Oct 22;9:484. doi: 10.3389/fgene.2018.00484. eCollection 2018.
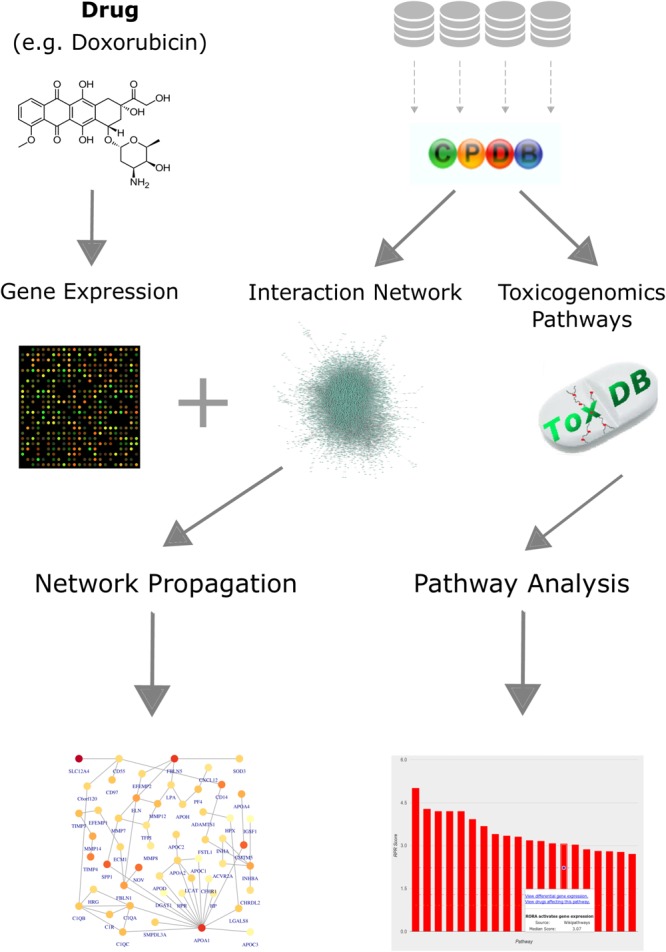
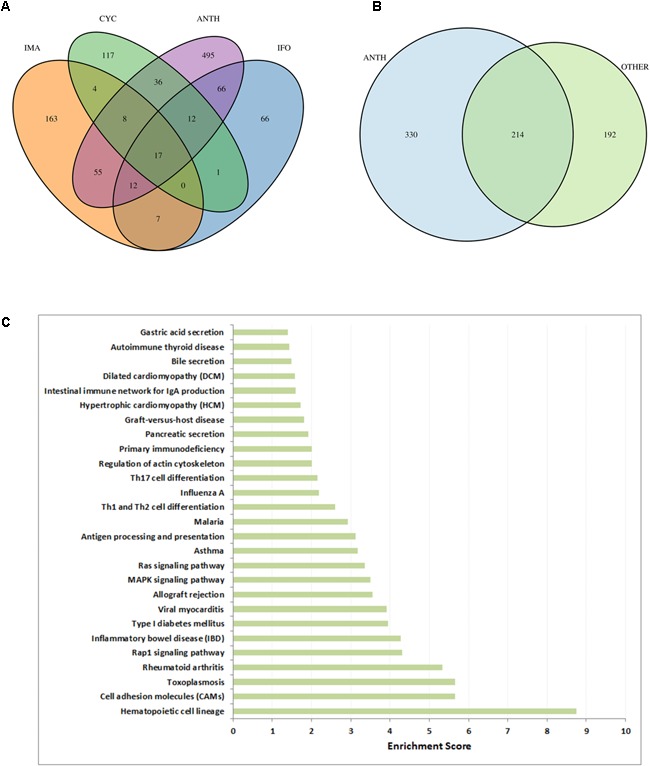
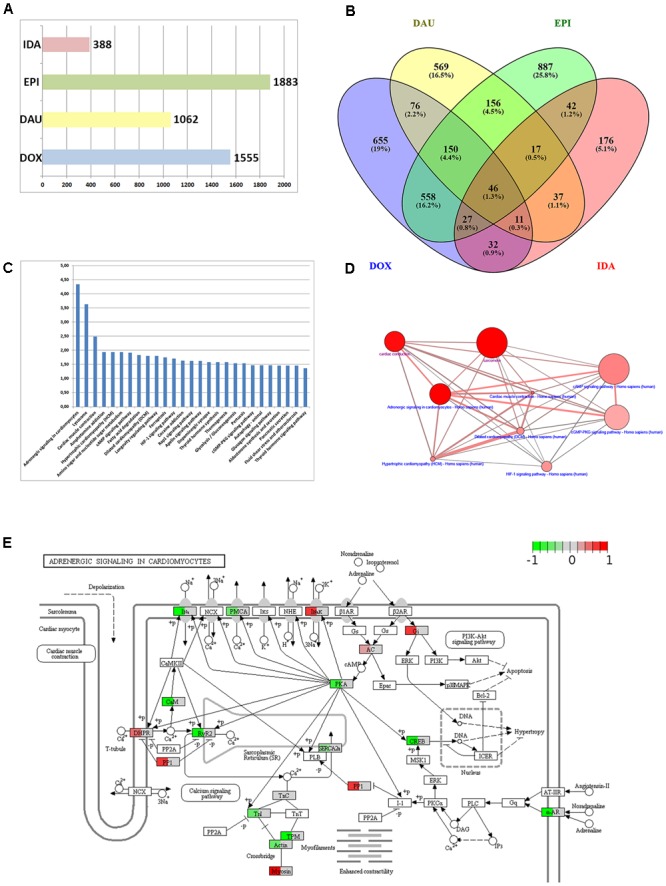
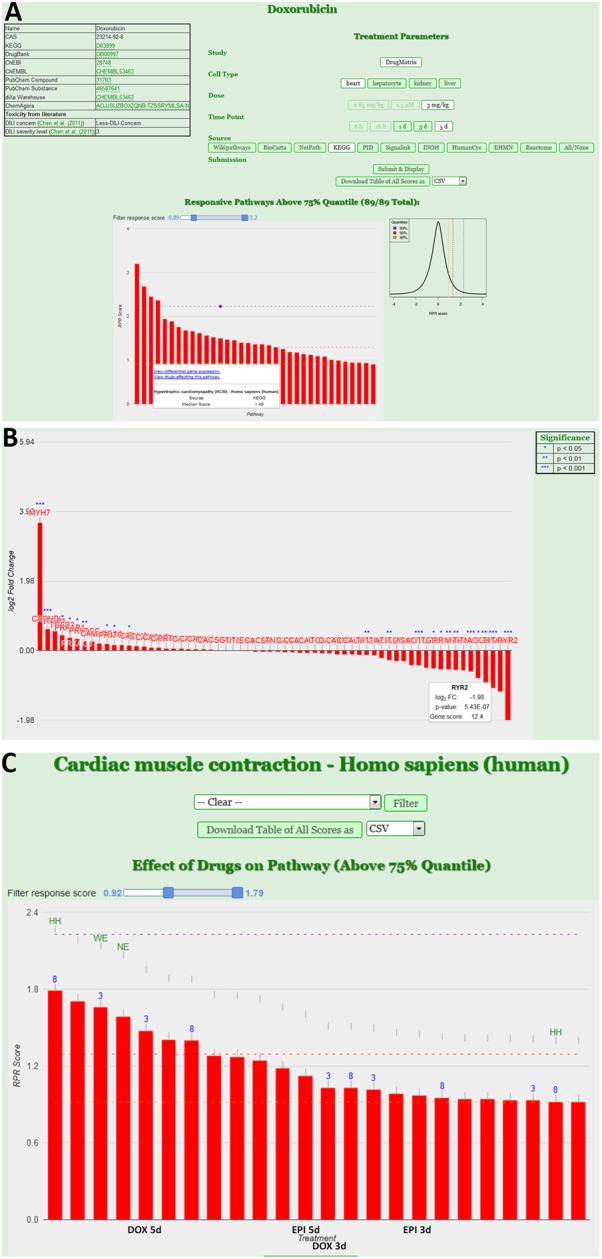
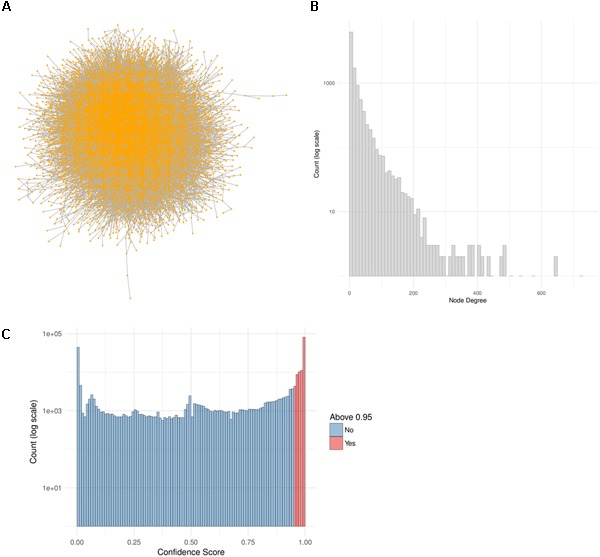
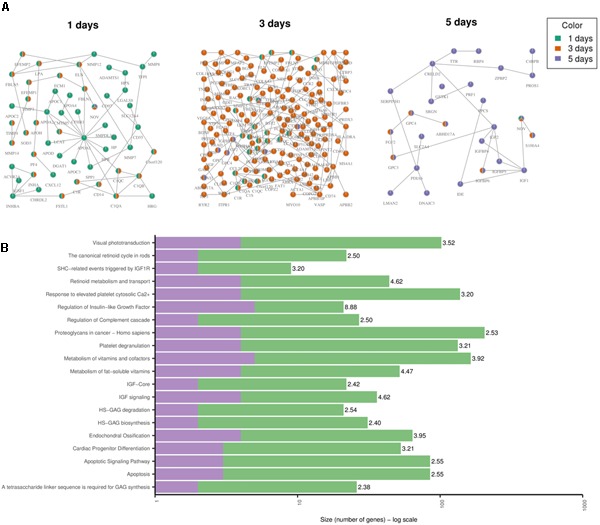
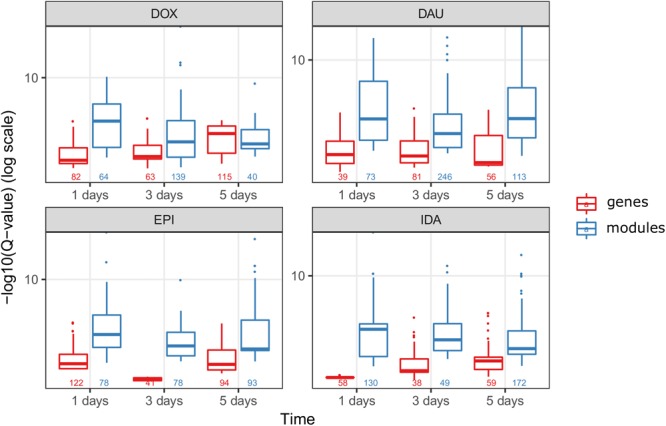
Toxicogenomics is the study of the molecular effects of chemical, biological and physical agents in biological systems, with the aim of elucidating toxicological mechanisms, building predictive models and improving diagnostics. The vast majority of toxicogenomics data has been generated at the transcriptome level, including RNA-seq and microarrays, and large quantities of drug-treatment data have been made publicly available through databases and repositories. Besides the identification of differentially expressed genes (DEGs) from case-control studies or drug treatment time series studies, bioinformatics methods have emerged that infer gene expression data at the molecular network and pathway level in order to reveal mechanistic information. In this work we describe different resources and tools that have been developed by us and others that relate gene expression measurements with known pathway information such as over-representation and gene set enrichment analyses. Furthermore, we highlight approaches that integrate gene expression data with molecular interaction networks in order to derive network modules related to drug toxicity. We describe the two main parts of the approach, i.e., the construction of a suitable molecular interaction network as well as the conduction of network propagation of the experimental data through the interaction network. In all cases we apply methods and tools to publicly available rat data on anthracyclines, an important class of anti-cancer drugs that are known to induce severe cardiotoxicity in patients. We report the results and functional implications achieved for four anthracyclines (doxorubicin, epirubicin, idarubicin, and daunorubicin) and compare the information content inherent in the different computational approaches.
毒理基因组学是研究化学、生物和物理因素在生物系统中的分子效应,旨在阐明毒理学机制、建立预测模型并改进诊断方法。绝大多数毒理基因组学数据是在转录组水平上生成的,包括RNA测序和微阵列,大量的药物治疗数据已通过数据库和储存库公开提供。除了从病例对照研究或药物治疗时间序列研究中识别差异表达基因(DEG)外,还出现了生物信息学方法,这些方法在分子网络和通路水平上推断基因表达数据,以揭示机制信息。在这项工作中,我们描述了我们自己和其他人开发的不同资源和工具,这些资源和工具将基因表达测量与已知的通路信息(如过度表达和基因集富集分析)联系起来。此外,我们强调了将基因表达数据与分子相互作用网络整合的方法,以获得与药物毒性相关的网络模块。我们描述了该方法的两个主要部分,即构建合适的分子相互作用网络以及通过相互作用网络对实验数据进行网络传播。在所有情况下,我们都将方法和工具应用于公开可用的大鼠蒽环类药物数据,蒽环类药物是一类重要的抗癌药物,已知会在患者中诱发严重的心脏毒性。我们报告了四种蒽环类药物(阿霉素、表柔比星、伊达比星和柔红霉素)的结果和功能意义,并比较了不同计算方法中固有的信息内容。