Selevsek Nathalie, Caiment Florian, Nudischer Ramona, Gmuender Hans, Agarkova Irina, Atkinson Francis L, Bachmann Ivo, Baier Vanessa, Barel Gal, Bauer Chris, Boerno Stefan, Bosc Nicolas, Clayton Olivia, Cordes Henrik, Deeb Sally, Gotta Stefano, Guye Patrick, Hersey Anne, Hunter Fiona M I, Kunz Laura, Lewalle Alex, Lienhard Matthias, Merken Jort, Minguet Jasmine, Oliveira Bernardo, Pluess Carla, Sarkans Ugis, Schrooders Yannick, Schuchhardt Johannes, Smit Ines, Thiel Christoph, Timmermann Bernd, Verheijen Marcha, Wittenberger Timo, Wolski Witold, Zerck Alexandra, Heymans Stephane, Kuepfer Lars, Roth Adrian, Schlapbach Ralph, Niederer Steven, Herwig Ralf, Kleinjans Jos
Functional Genomics Center, ETH Zurich, Switzerland.
Department of Toxicogenomics, Maastricht University, Maastricht, The Netherlands.
Commun Biol. 2020 Oct 15;3(1):573. doi: 10.1038/s42003-020-01302-8.
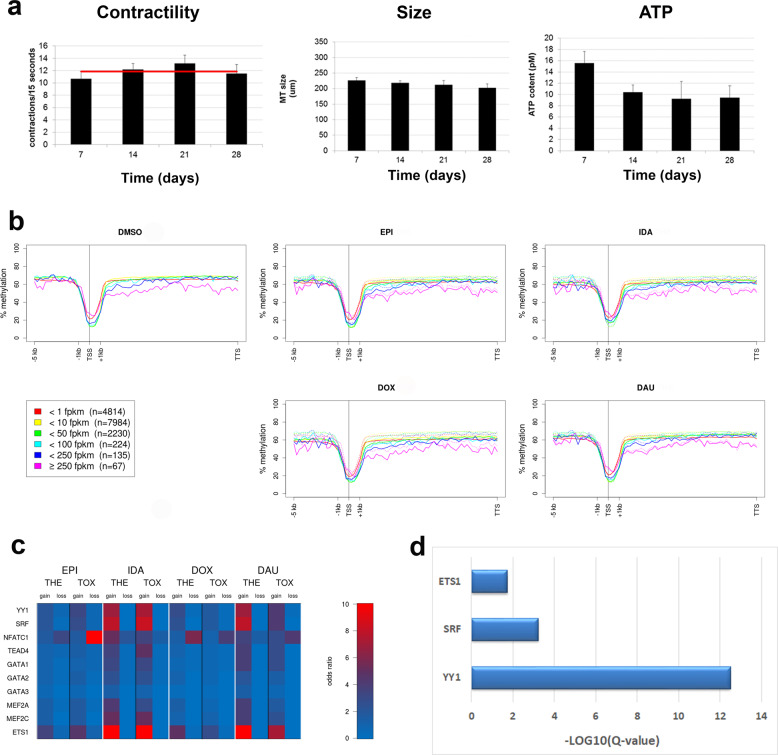
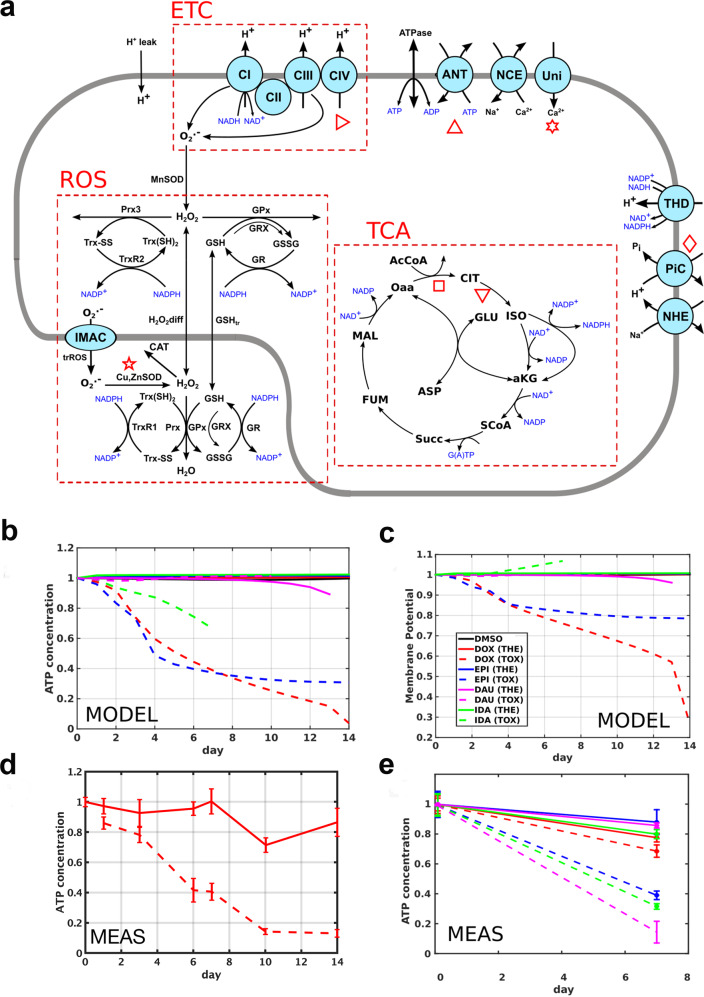
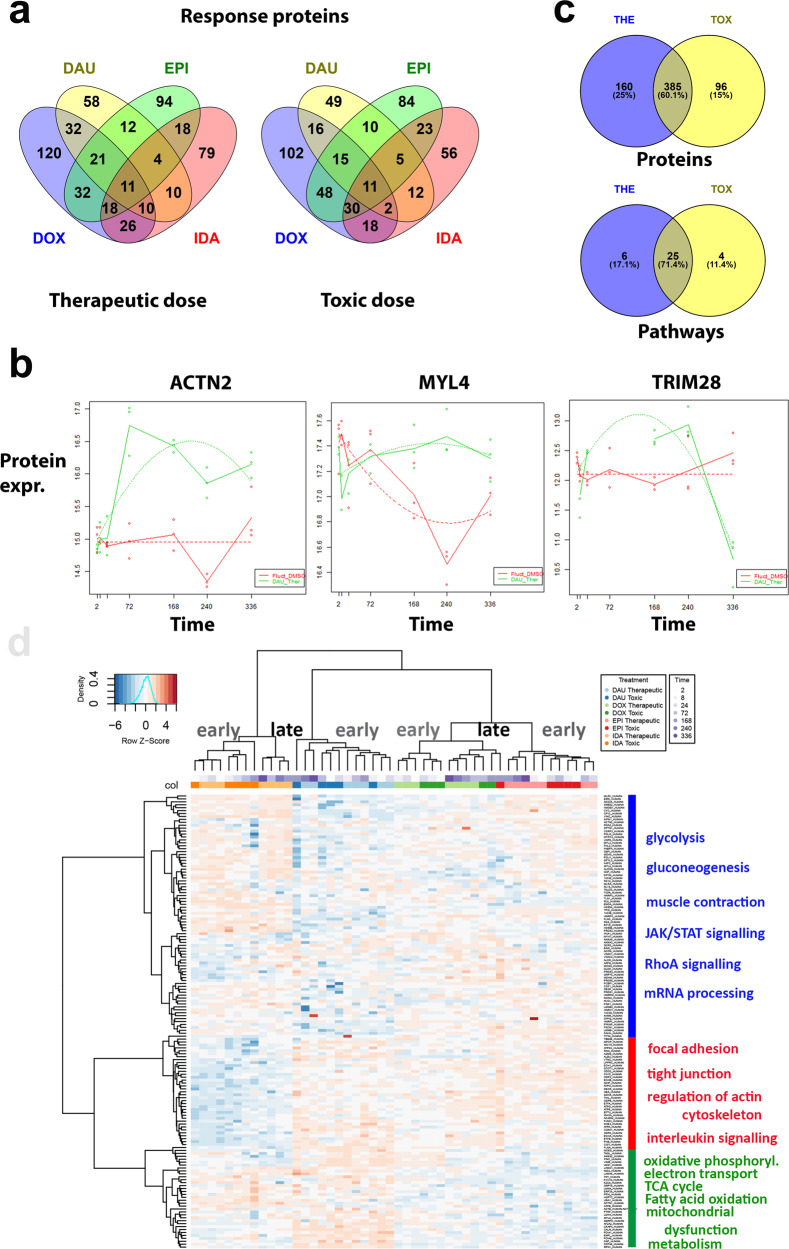
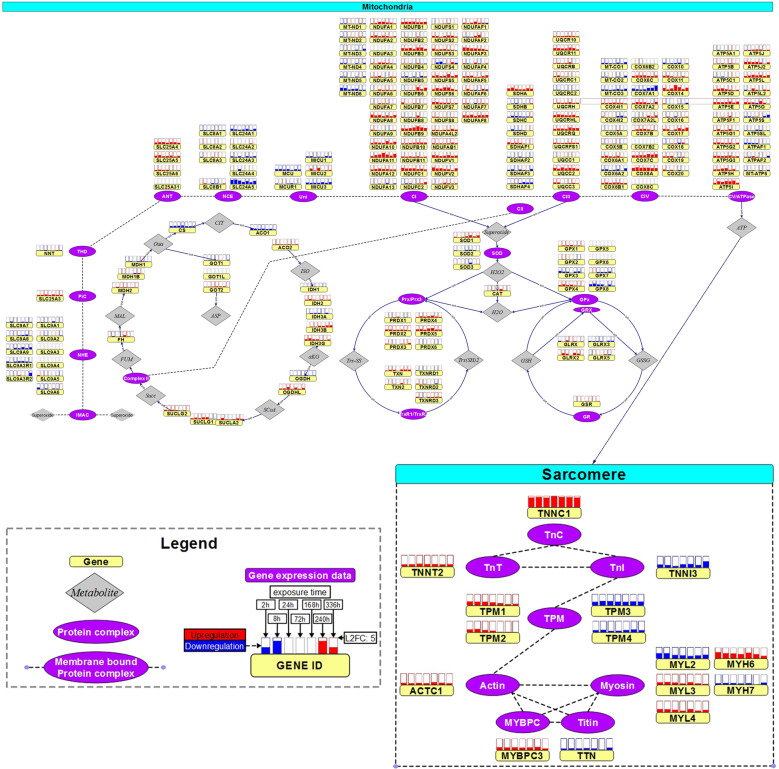
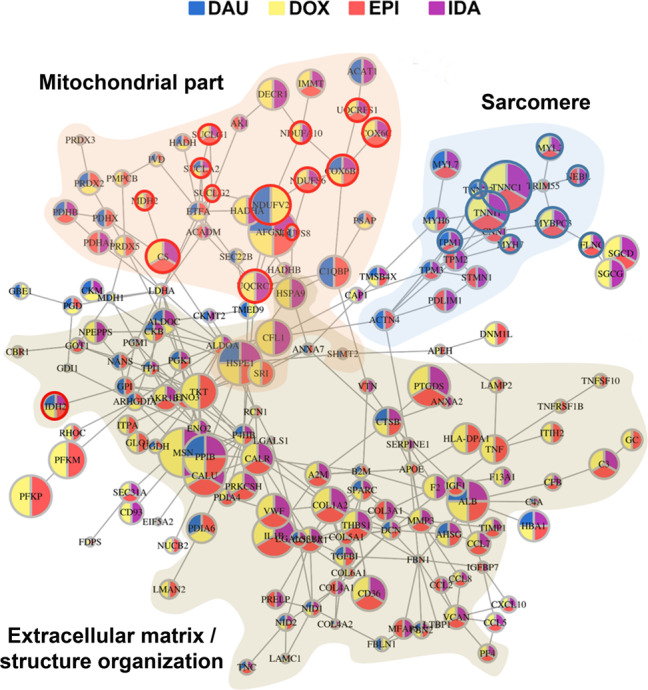
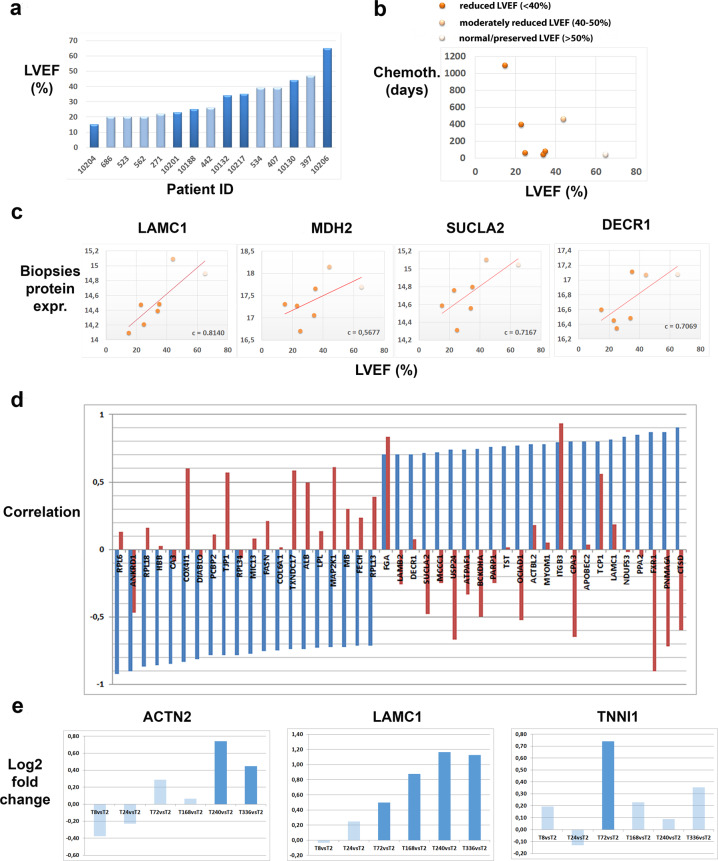
Uncovering cellular responses from heterogeneous genomic data is crucial for molecular medicine in particular for drug safety. This can be realized by integrating the molecular activities in networks of interacting proteins. As proof-of-concept we challenge network modeling with time-resolved proteome, transcriptome and methylome measurements in iPSC-derived human 3D cardiac microtissues to elucidate adverse mechanisms of anthracycline cardiotoxicity measured with four different drugs (doxorubicin, epirubicin, idarubicin and daunorubicin). Dynamic molecular analysis at in vivo drug exposure levels reveal a network of 175 disease-associated proteins and identify common modules of anthracycline cardiotoxicity in vitro, related to mitochondrial and sarcomere function as well as remodeling of extracellular matrix. These in vitro-identified modules are transferable and are evaluated with biopsies of cardiomyopathy patients. This to our knowledge most comprehensive study on anthracycline cardiotoxicity demonstrates a reproducible workflow for molecular medicine and serves as a template for detecting adverse drug responses from complex omics data.
从异质基因组数据中揭示细胞反应对于分子医学尤其是药物安全性至关重要。这可以通过整合相互作用蛋白质网络中的分子活性来实现。作为概念验证,我们用诱导多能干细胞衍生的人类3D心脏微组织中的时间分辨蛋白质组、转录组和甲基化组测量对网络建模提出挑战,以阐明用四种不同药物(阿霉素、表柔比星、伊达比星和柔红霉素)测量的蒽环类药物心脏毒性的不良机制。在体内药物暴露水平下的动态分子分析揭示了一个由175种疾病相关蛋白质组成的网络,并在体外确定了蒽环类药物心脏毒性的常见模块,这些模块与线粒体和肌节功能以及细胞外基质重塑有关。这些在体外确定的模块具有可转移性,并通过心肌病患者的活检进行评估。据我们所知,这项关于蒽环类药物心脏毒性的最全面研究展示了分子医学中一种可重复的工作流程,并作为从复杂组学数据中检测药物不良反应的模板。