bioMérieux, Marcy l'Étoile, France.
Univ Lyon, Université Lyon 1, CNRS, Laboratoire de Biométrie et Biologie Evolutive UMR5558 F-69622 Villeurbanne, France.
PLoS Genet. 2018 Nov 12;14(11):e1007758. doi: 10.1371/journal.pgen.1007758. eCollection 2018 Nov.
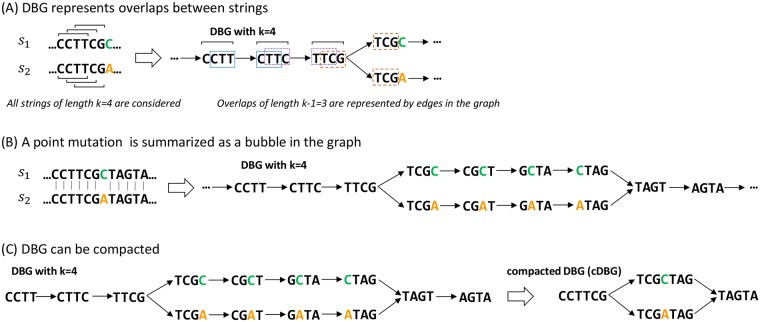
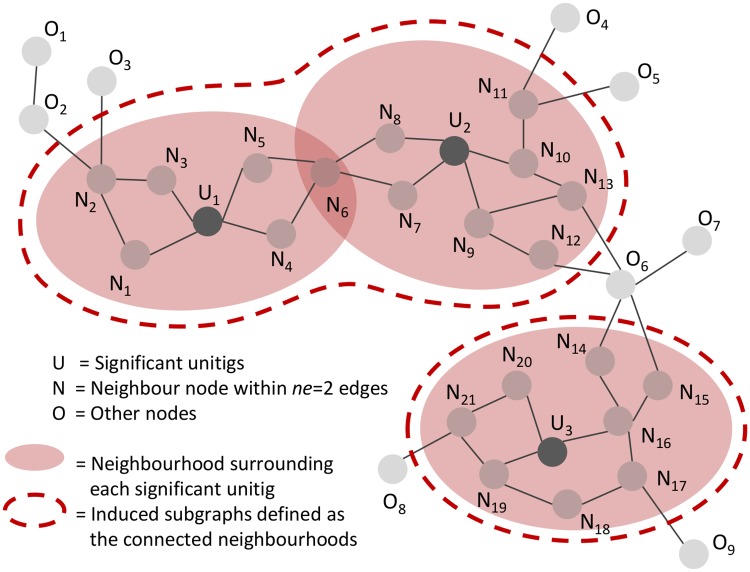
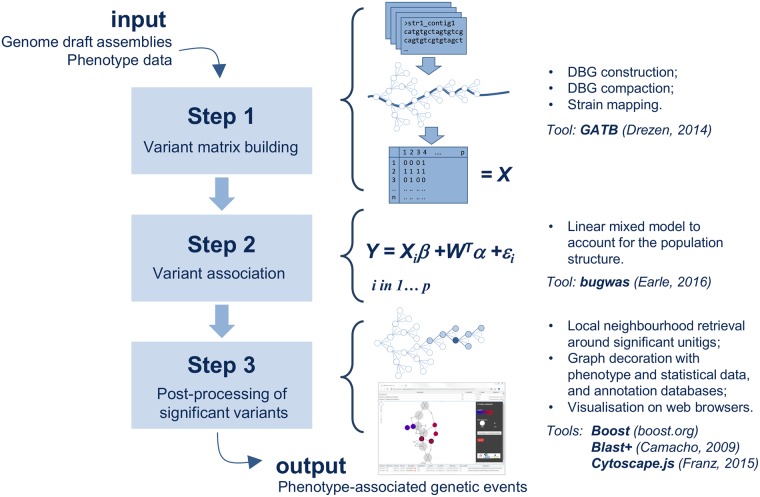
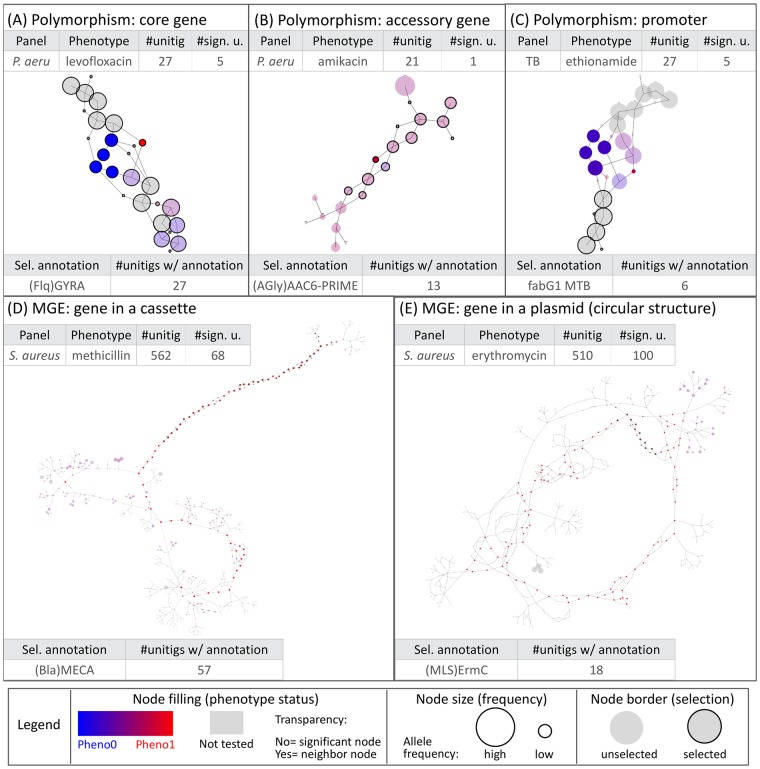
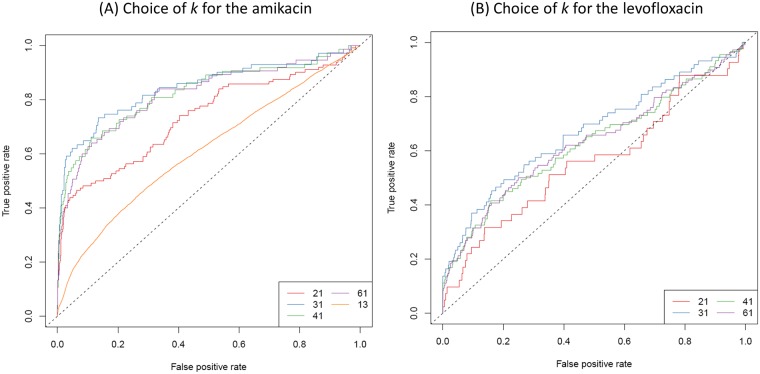
Genome-wide association study (GWAS) methods applied to bacterial genomes have shown promising results for genetic marker discovery or detailed assessment of marker effect. Recently, alignment-free methods based on k-mer composition have proven their ability to explore the accessory genome. However, they lead to redundant descriptions and results which are sometimes hard to interpret. Here we introduce DBGWAS, an extended k-mer-based GWAS method producing interpretable genetic variants associated with distinct phenotypes. Relying on compacted De Bruijn graphs (cDBG), our method gathers cDBG nodes, identified by the association model, into subgraphs defined from their neighbourhood in the initial cDBG. DBGWAS is alignment-free and only requires a set of contigs and phenotypes. In particular, it does not require prior annotation or reference genomes. It produces subgraphs representing phenotype-associated genetic variants such as local polymorphisms and mobile genetic elements (MGE). It offers a graphical framework which helps interpret GWAS results. Importantly it is also computationally efficient-experiments took one hour and a half on average. We validated our method using antibiotic resistance phenotypes for three bacterial species. DBGWAS recovered known resistance determinants such as mutations in core genes in Mycobacterium tuberculosis, and genes acquired by horizontal transfer in Staphylococcus aureus and Pseudomonas aeruginosa-along with their MGE context. It also enabled us to formulate new hypotheses involving genetic variants not yet described in the antibiotic resistance literature. An open-source tool implementing DBGWAS is available at https://gitlab.com/leoisl/dbgwas.
全基因组关联研究(GWAS)方法应用于细菌基因组,已显示出在遗传标记发现或详细评估标记效应方面的有前景的结果。最近,基于 k-mer 组成的无比对方法已证明其探索附属基因组的能力。然而,它们导致了冗余的描述和结果,有时难以解释。在这里,我们介绍了 DBGWAS,这是一种扩展的基于 k-mer 的 GWAS 方法,可产生与不同表型相关的可解释遗传变异。该方法依赖于紧凑的布劳因图(cDBG),通过关联模型识别 cDBG 节点,并将其聚集到由初始 cDBG 中的邻近节点定义的子图中。DBGWAS 是无比对的,只需要一组 contigs 和表型。特别是,它不需要预先注释或参考基因组。它生成表示与表型相关的遗传变异的子图,例如局部多态性和移动遗传元件(MGE)。它提供了一个图形框架,有助于解释 GWAS 结果。重要的是,它的计算效率也很高,实验平均需要一个半小时。我们使用三种细菌的抗生素耐药表型验证了我们的方法。DBGWAS 恢复了已知的耐药决定因素,如结核分枝杆菌核心基因中的突变,以及金黄色葡萄球菌和铜绿假单胞菌中水平转移获得的基因及其 MGE 背景。它还使我们能够提出新的假设,涉及尚未在抗生素耐药性文献中描述的遗传变异。一个实现 DBGWAS 的开源工具可在 https://gitlab.com/leoisl/dbgwas 上获得。