Centro de Investigación en Enfermedades Tropicales, Facultad de Microbiología, Universidad de Costa Rica, San José, Costa Rica.
Centro de Investigación en Biología Celular y Molecular, Facultad de Microbiología, Universidad de Costa Rica, San José, Costa Rica.
Sci Rep. 2020 Jan 29;10(1):1392. doi: 10.1038/s41598-020-58319-6.
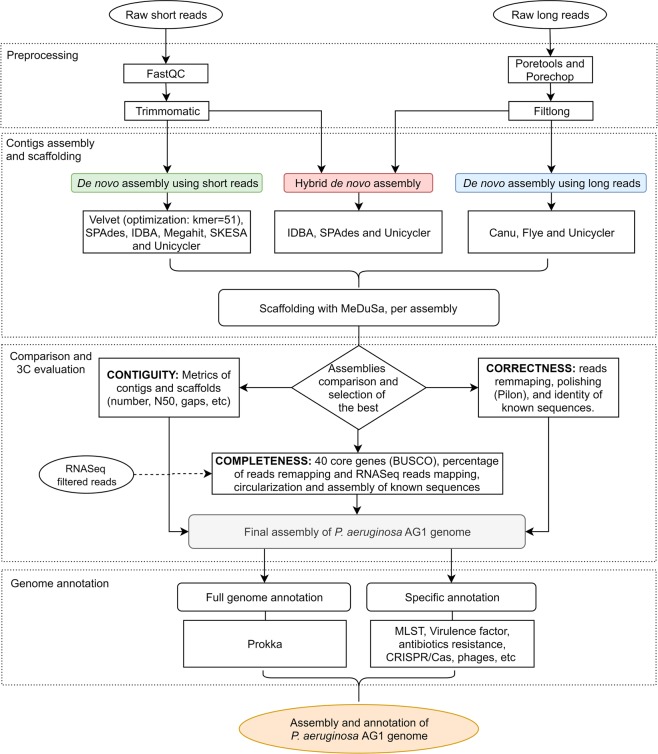
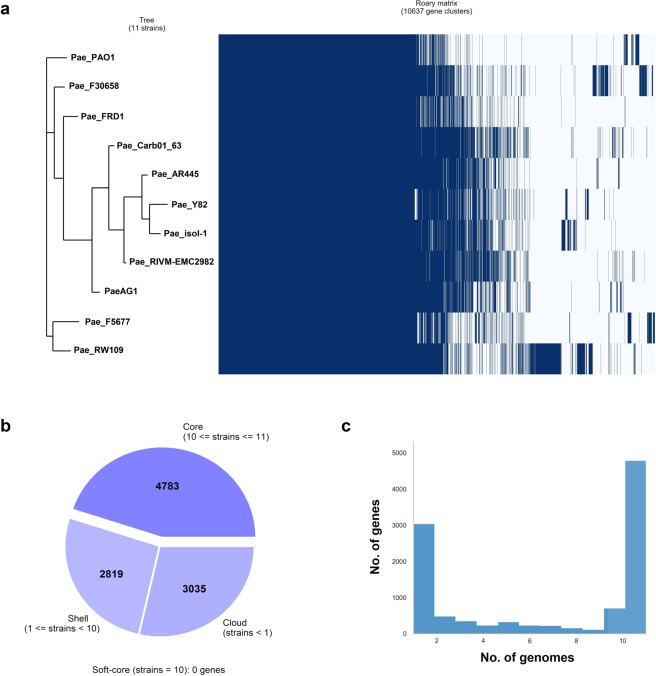
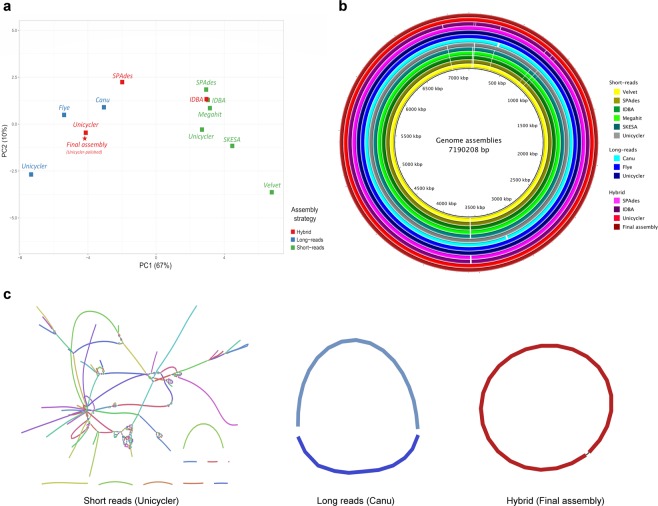
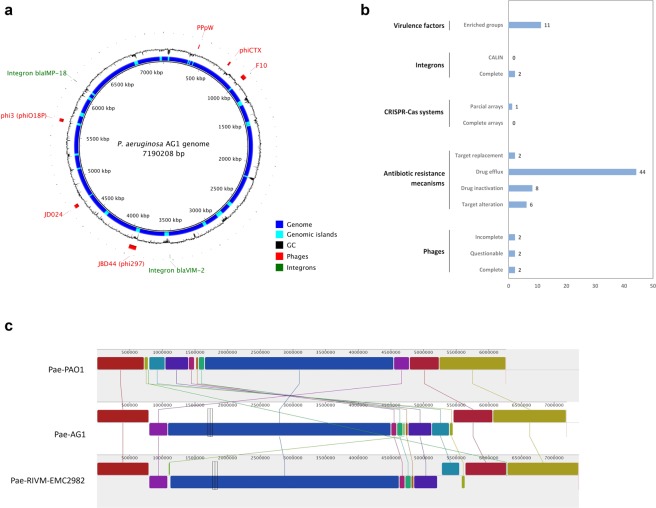
Genotyping methods and genome sequencing are indispensable to reveal genomic structure of bacterial species displaying high level of genome plasticity. However, reconstruction of genome or assembly is not straightforward due to data complexity, including repeats, mobile and accessory genetic elements of bacterial genomes. Moreover, since the solution to this problem is strongly influenced by sequencing technology, bioinformatics pipelines, and selection criteria to assess assemblers, there is no systematic way to select a priori the optimal assembler and parameter settings. To assembly the genome of Pseudomonas aeruginosa strain AG1 (PaeAG1), short reads (Illumina) and long reads (Oxford Nanopore) sequencing data were used in 13 different non-hybrid and hybrid approaches. PaeAG1 is a multiresistant high-risk sequence type 111 (ST-111) clone that was isolated from a Costa Rican hospital and it was the first report of an isolate of P. aeruginosa carrying both blaVIM-2 and blaIMP-18 genes encoding for metallo-β-lactamases (MBL) enzymes. To assess the assemblies, multiple metrics regard to contiguity, correctness and completeness (3C criterion, as we define here) were used for benchmarking the 13 approaches and select a definitive assembly. In addition, annotation was done to identify genes (coding and RNA regions) and to describe the genomic content of PaeAG1. Whereas long reads and hybrid approaches showed better performances in terms of contiguity, higher correctness and completeness metrics were obtained for short read only and hybrid approaches. A manually curated and polished hybrid assembly gave rise to a single circular sequence with 100% of core genes and known regions identified, >98% of reads mapped back, no gaps, and uniform coverage. The strategy followed to obtain this high-quality 3C assembly is detailed in the manuscript and we provide readers with an all-in-one script to replicate our results or to apply it to other troublesome cases. The final 3C assembly revealed that the PaeAG1 genome has 7,190,208 bp, a 65.7% GC content and 6,709 genes (6,620 coding sequences), many of which are included in multiple mobile genomic elements, such as 57 genomic islands, six prophages, and two complete integrons with blaVIM-2 and blaIMP-18 MBL genes. Up to 250 and 60 of the predicted genes are anticipated to play a role in virulence (adherence, quorum sensing and secretion) or antibiotic resistance (β-lactamases, efflux pumps, etc). Altogether, the assembly and annotation of the PaeAG1 genome provide new perspectives to continue studying the genomic diversity and gene content of this important human pathogen.
基因分型方法和基因组测序对于揭示具有高水平基因组可塑性的细菌物种的基因组结构是不可或缺的。然而,由于数据的复杂性,包括细菌基因组中的重复、移动和辅助遗传元件,基因组的重建或组装并不简单。此外,由于这个问题的解决方案受到测序技术、生物信息学管道以及评估组装器的选择标准的强烈影响,因此没有系统的方法可以预先选择最佳的组装器和参数设置。
为了组装铜绿假单胞菌 AG1 菌株(PaeAG1)的基因组,使用了 13 种不同的非杂交和杂交方法的短读(Illumina)和长读(Oxford Nanopore)测序数据。PaeAG1 是一种多耐药高风险序列类型 111(ST-111)克隆,它是从哥斯达黎加的一家医院分离出来的,这是首次报告携带 blaVIM-2 和 blaIMP-18 基因编码金属β-内酰胺酶(MBL)酶的铜绿假单胞菌分离株。为了评估组装结果,我们使用了多种关于连续性、正确性和完整性的指标(我们在这里定义为 3C 标准)来对 13 种方法进行基准测试,并选择了一个确定的组装。此外,我们还进行了注释,以识别基因(编码和 RNA 区域)并描述 PaeAG1 的基因组内容。尽管长读和杂交方法在连续性方面表现更好,但仅使用短读和杂交方法可以获得更高的正确性和完整性指标。经过精心编辑和润色的杂交组装得到了一个单一的圆形序列,其中包含 100%的核心基因和已知区域,>98%的reads 可以映射回,没有缺口,并且覆盖均匀。本文详细介绍了获得这种高质量 3C 组装的策略,并为读者提供了一个完整的脚本,以复制我们的结果或将其应用于其他棘手的情况。最终的 3C 组装结果表明,PaeAG1 基因组有 7,190,208bp,GC 含量为 65.7%,有 6,709 个基因(6,620 个编码序列),其中许多基因都包含在多个移动基因组元件中,如 57 个基因组岛、6 个噬菌体和两个完整的整合子,其中包含 blaVIM-2 和 blaIMP-18 MBL 基因。预计多达 250 个和 60 个预测基因将在毒力(粘附、群体感应和分泌)或抗生素耐药性(β-内酰胺酶、外排泵等)中发挥作用。总之,PaeAG1 基因组的组装和注释为继续研究这种重要的人类病原体的基因组多样性和基因内容提供了新的视角。