Cheng Qing, Song Sang-Ho, Augustine George J
Laboratory of Neurobiology, National Institute of Environmental Health Sciences, National Institutes of Health, Durham, NC, United States.
Lee Kong Chian School of Medicine, Nanyang Technological University, Singapore, Singapore.
Front Synaptic Neurosci. 2018 Oct 30;10:33. doi: 10.3389/fnsyn.2018.00033. eCollection 2018.
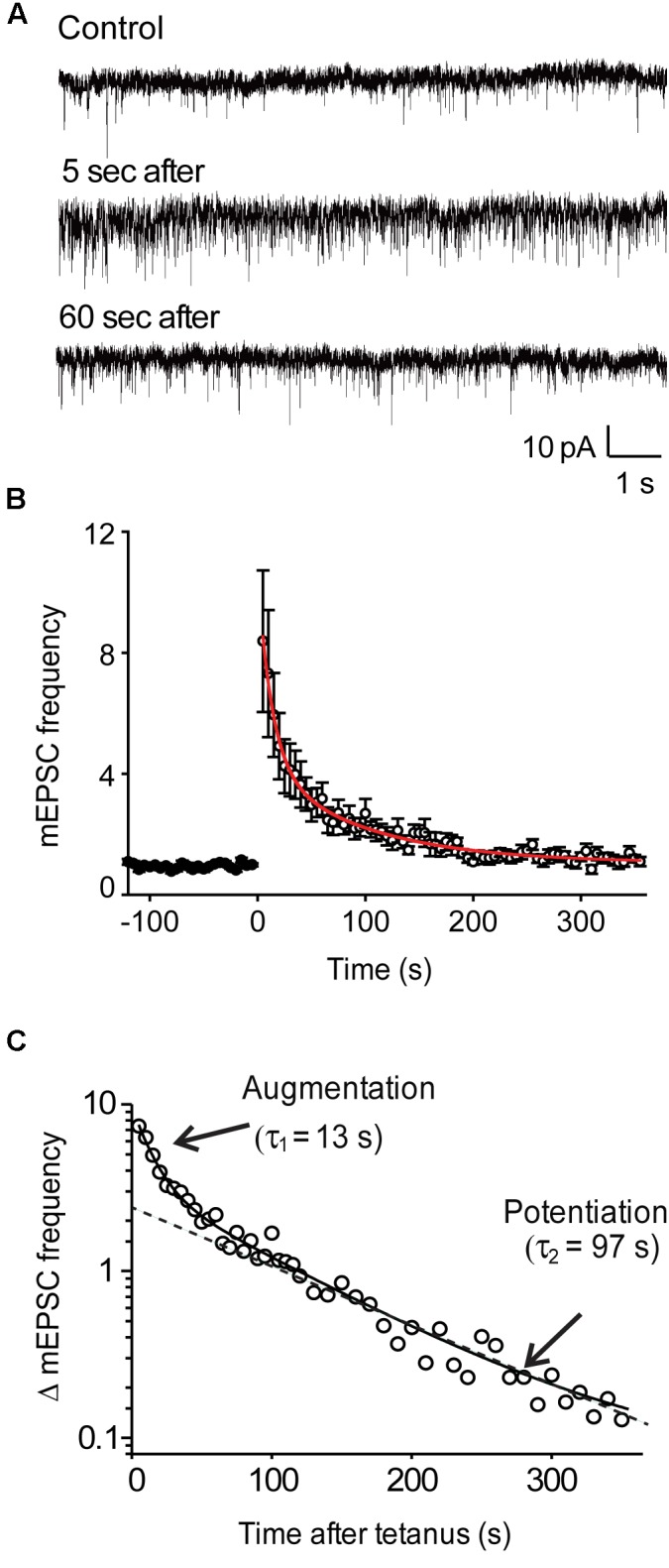
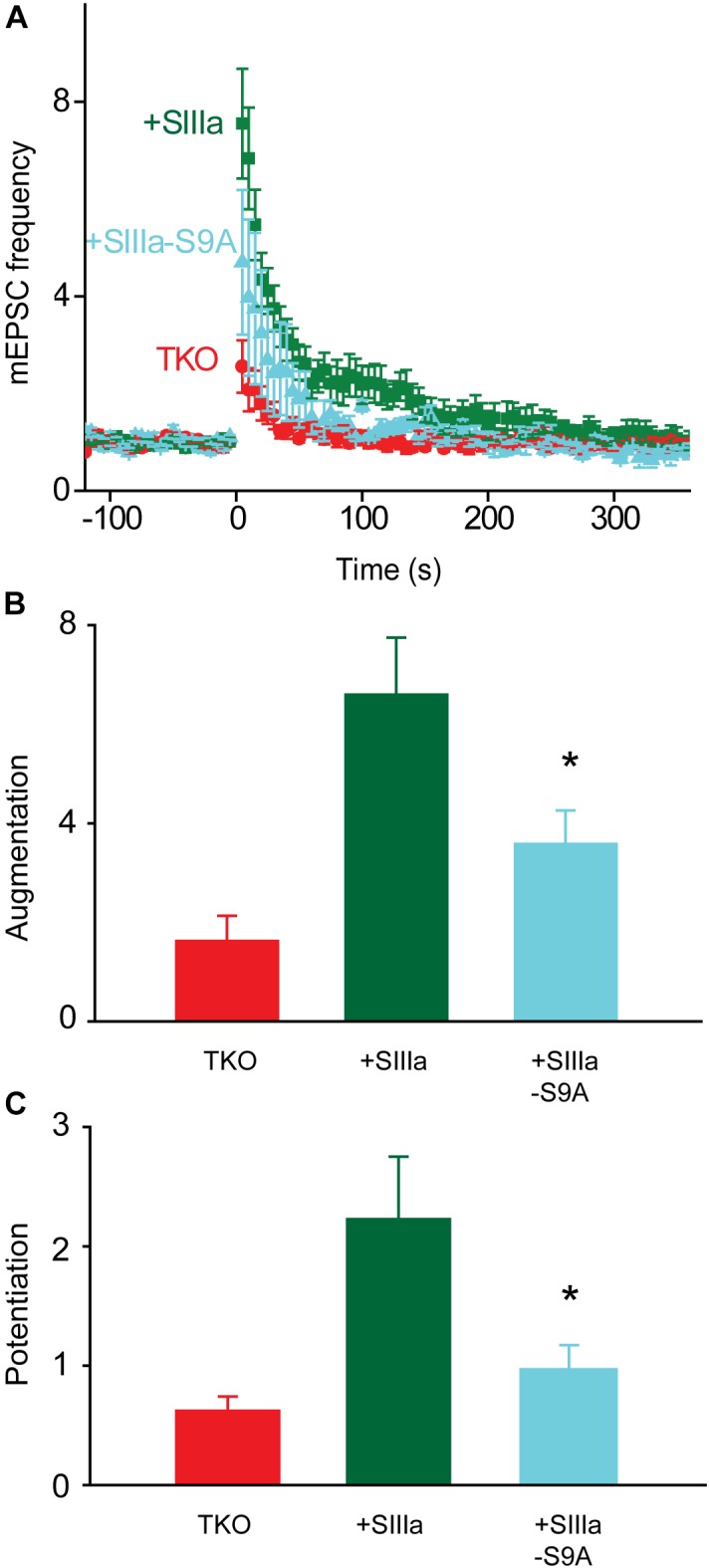
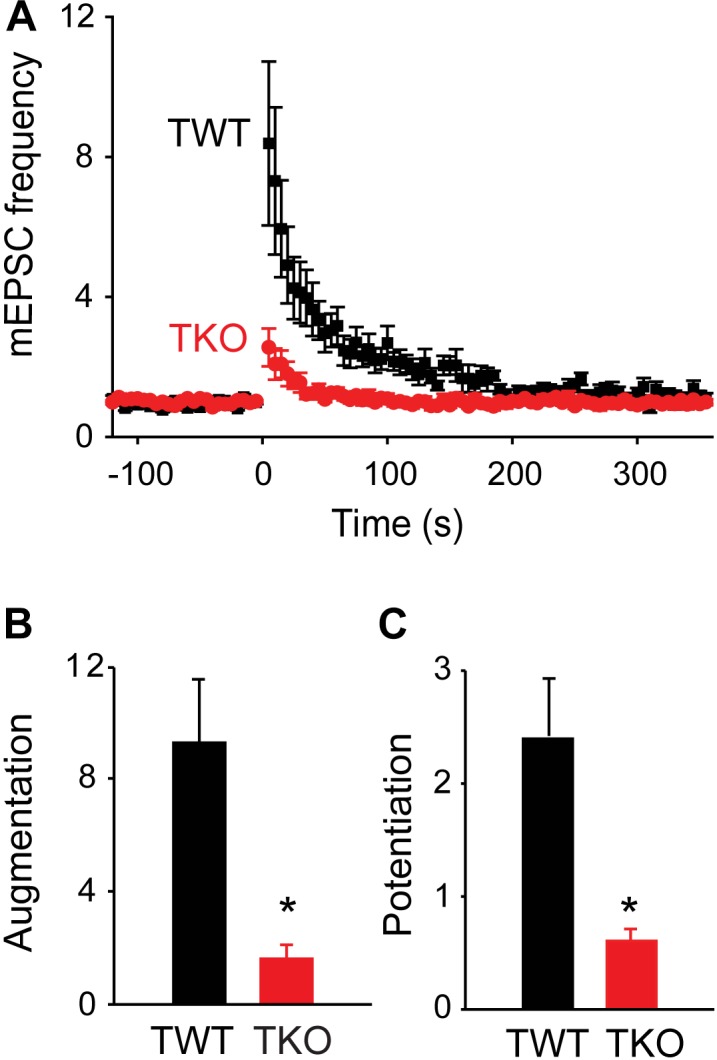
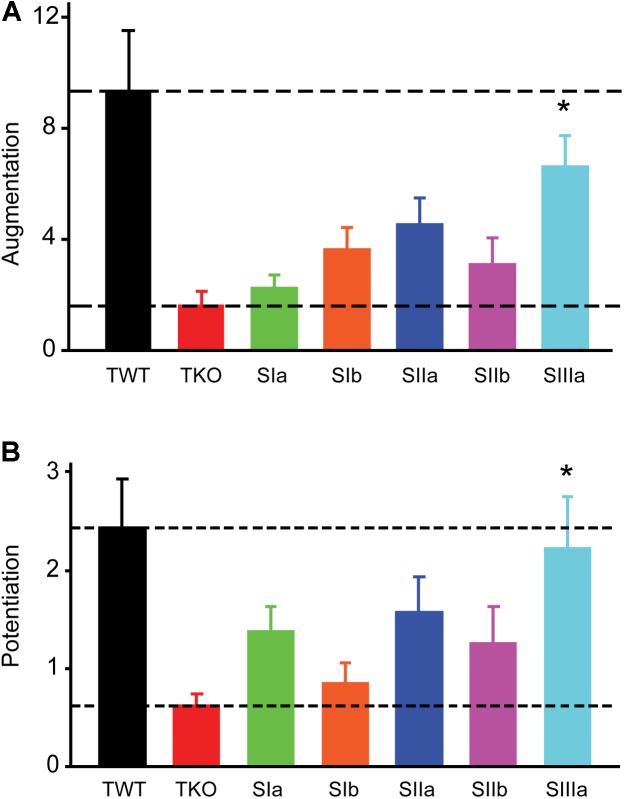
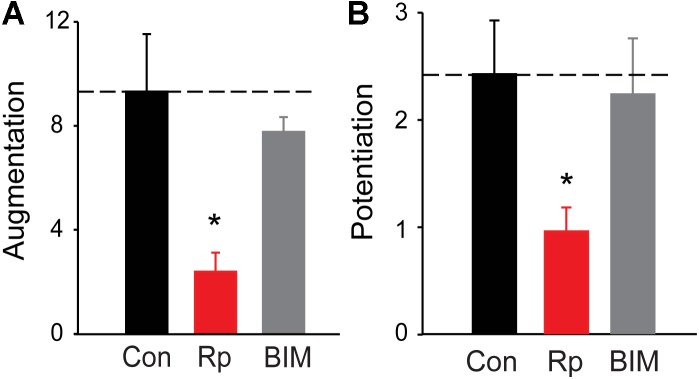
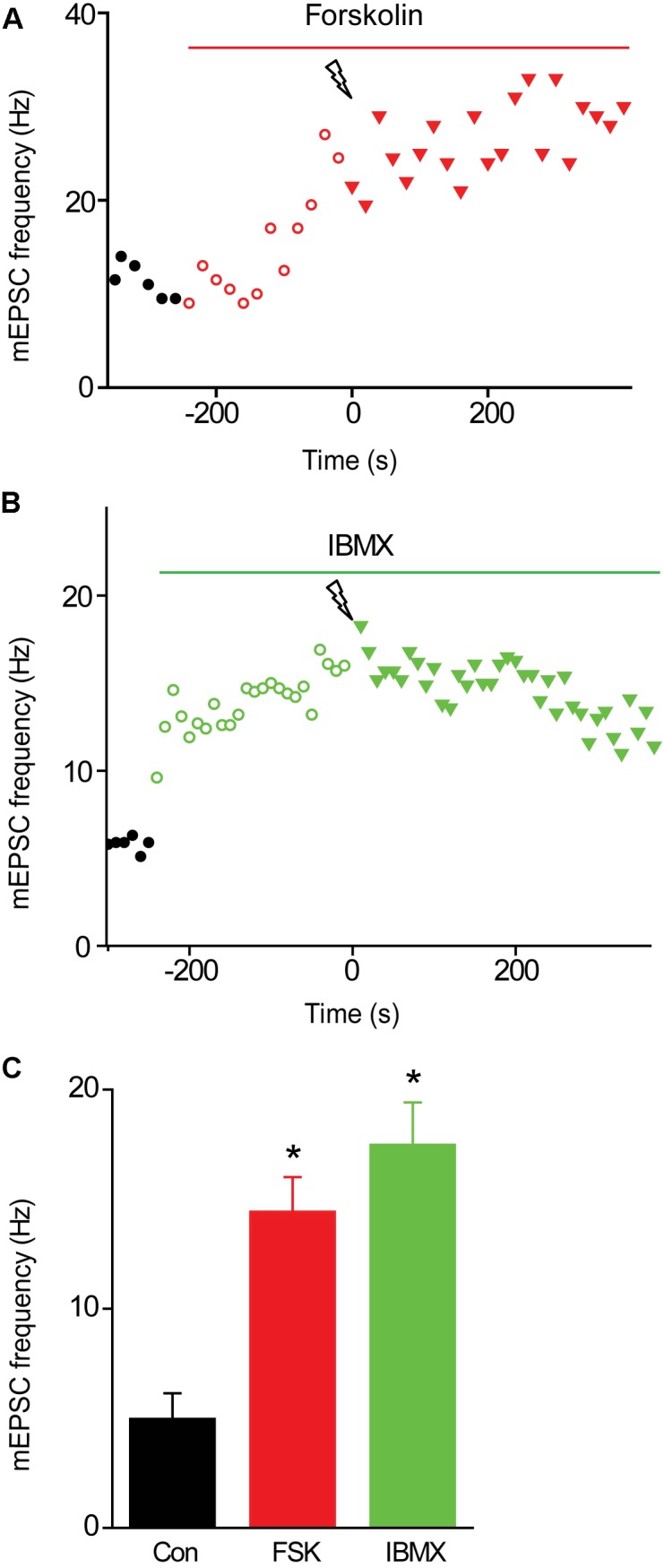
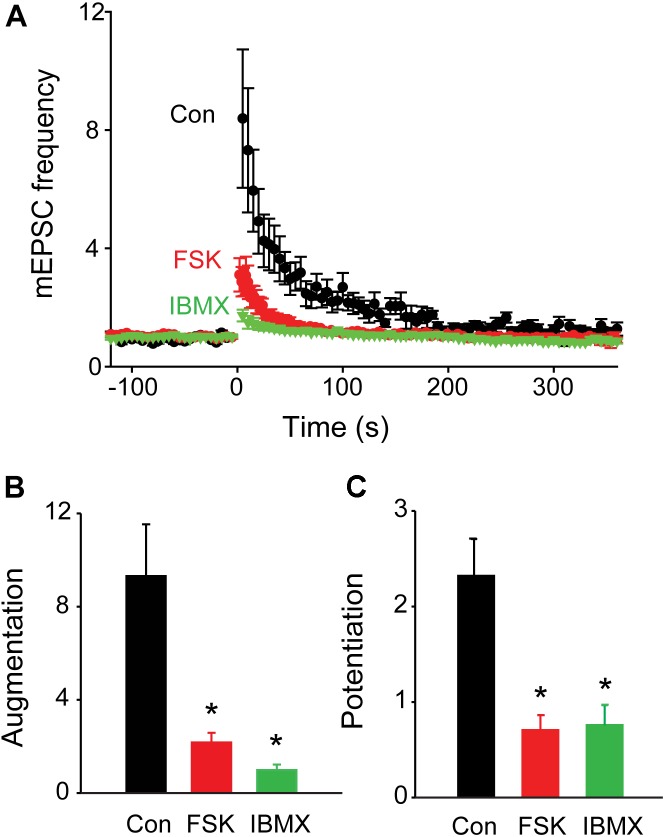
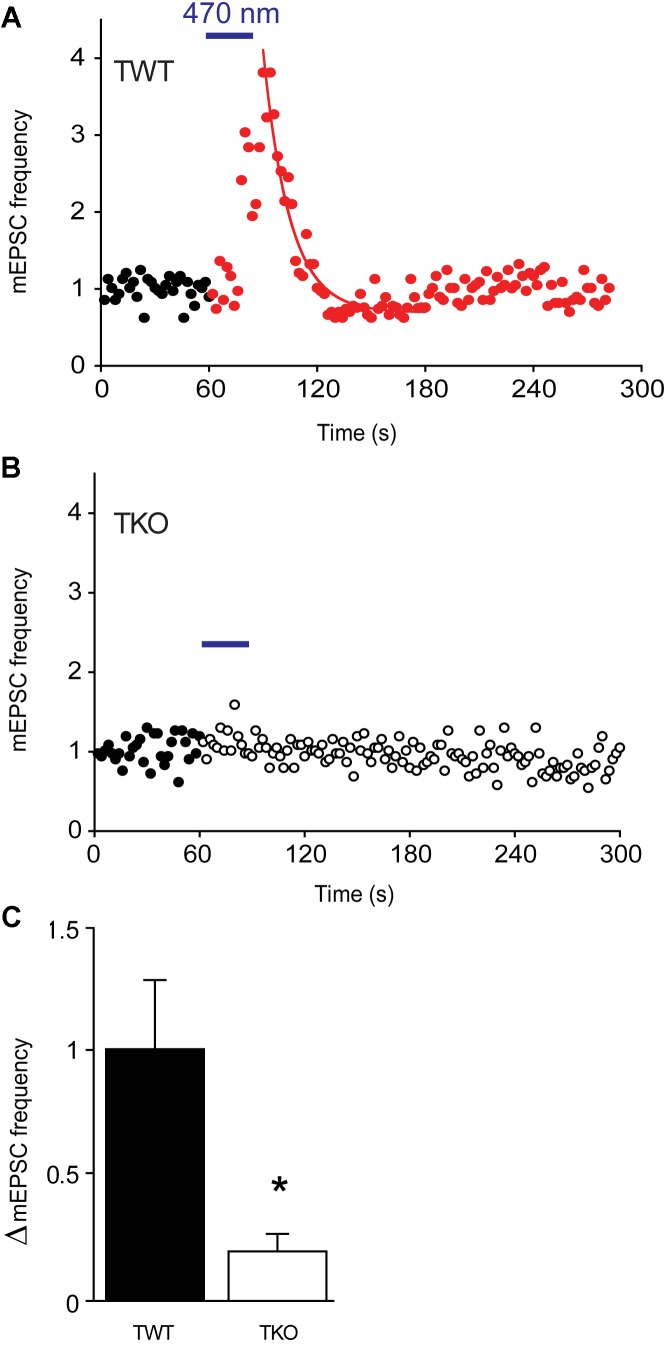
We used genetic and pharmacological approaches to identify the signaling pathways involved in augmentation and potentiation, two forms of activity dependent, short-term synaptic plasticity that enhance neurotransmitter release. Trains of presynaptic action potentials produced a robust increase in the frequency of miniature excitatory postsynaptic currents (mEPSCs). Following the end of the stimulus, mEPSC frequency followed a bi-exponential decay back to basal levels. The time constants of decay identified these two exponential components as the decay of augmentation and potentiation, respectively. Augmentation increased mEPSC frequency by 9.3-fold, while potentiation increased mEPSC frequency by 2.4-fold. In synapsin triple-knockout (TKO) neurons, augmentation was reduced by 83% and potentiation was reduced by 74%, suggesting that synapsins are key signaling elements in both forms of plasticity. To examine the synapsin isoforms involved, we expressed individual synapsin isoforms in TKO neurons. While synapsin IIIa rescued both augmentation and potentiation, none of the other synapsin isoforms produced statistically significant amounts of rescue. To determine the involvement of protein kinases in these two forms of short-term plasticity, we examined the effects of inhibitors of protein kinases A (PKA) and C (PKC). While inhibition of PKC had little effect, PKA inhibition reduced augmentation by 76% and potentiation by 60%. Further, elevation of intracellular cAMP concentration, by either forskolin or IBMX, greatly increased mEPSC frequency and occluded the amount of augmentation and potentiation evoked by electrical stimulation. Finally, mutating a PKA phosphorylation site to non-phosphorylatable alanine largely abolished the ability of synapsin IIIa to rescue both augmentation and potentiation. Together, these results indicate that PKA activation is required for both augmentation and potentiation of spontaneous neurotransmitter release and that PKA-mediated phosphorylation of synapsin IIIa underlies both forms of presynaptic short-term plasticity.
我们采用遗传学和药理学方法来确定参与增强和易化作用的信号通路,这是两种依赖于活动的短期突触可塑性形式,可增强神经递质释放。一连串的突触前动作电位使微小兴奋性突触后电流(mEPSCs)的频率显著增加。刺激结束后,mEPSC频率呈双指数衰减回到基础水平。衰减的时间常数将这两个指数成分分别确定为增强和易化作用的衰减。增强作用使mEPSC频率增加了9.3倍,而易化作用使mEPSC频率增加了2.4倍。在突触结合蛋白三敲除(TKO)神经元中,增强作用降低了83%,易化作用降低了74%,这表明突触结合蛋白是这两种可塑性形式中的关键信号元件。为了研究涉及的突触结合蛋白亚型,我们在TKO神经元中表达了单个突触结合蛋白亚型。虽然突触结合蛋白IIIa挽救了增强和易化作用,但其他突触结合蛋白亚型均未产生具有统计学意义的挽救效果。为了确定蛋白激酶在这两种短期可塑性形式中的作用,我们研究了蛋白激酶A(PKA)和C(PKC)抑制剂的影响。虽然抑制PKC影响不大,但抑制PKA使增强作用降低了76%,易化作用降低了60%。此外,通过福斯可林或异丁基甲基黄嘌呤提高细胞内cAMP浓度,可大大增加mEPSC频率,并消除电刺激诱发的增强和易化作用量。最后,将一个PKA磷酸化位点突变为不可磷酸化的丙氨酸,在很大程度上消除了突触结合蛋白IIIa挽救增强和易化作用的能力。总之,这些结果表明,PKA激活是自发神经递质释放增强和易化作用所必需的,并且PKA介导的突触结合蛋白IIIa磷酸化是两种形式的突触前短期可塑性的基础。