Department of Molecular Medicine and Medical Biotechnology, School of Medicine, University of Naples Federico II, 80131 Naples, Italy.
CEINGE-Biotecnologie Avanzate s.c.ar.l., 80145 Naples, Italy.
Int J Mol Sci. 2020 Jul 15;21(14):4998. doi: 10.3390/ijms21144998.
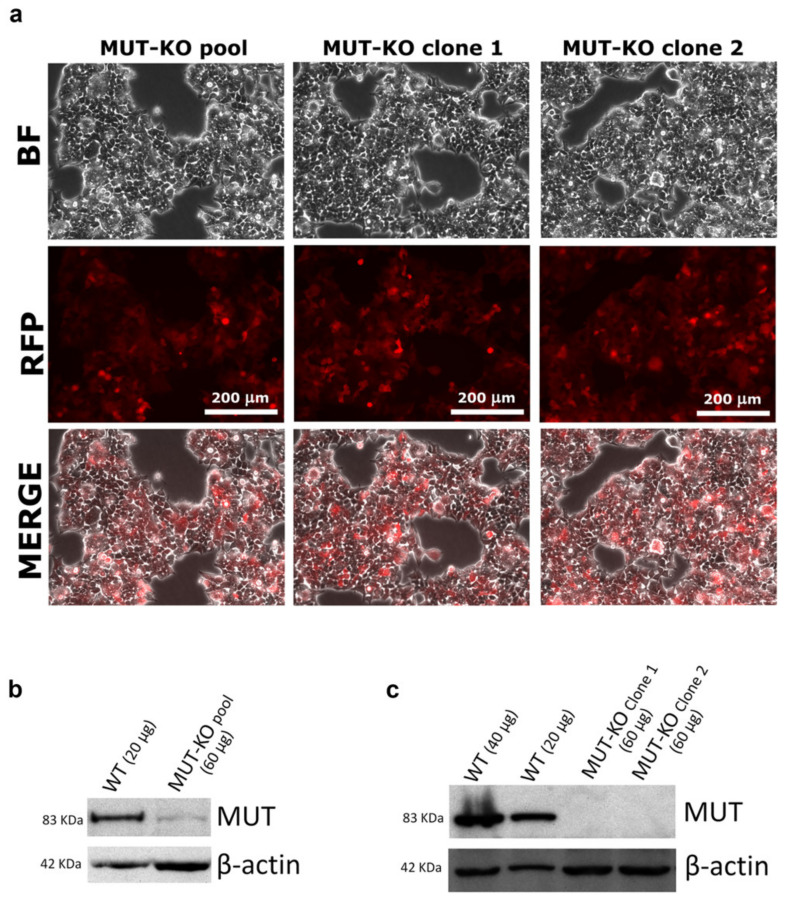
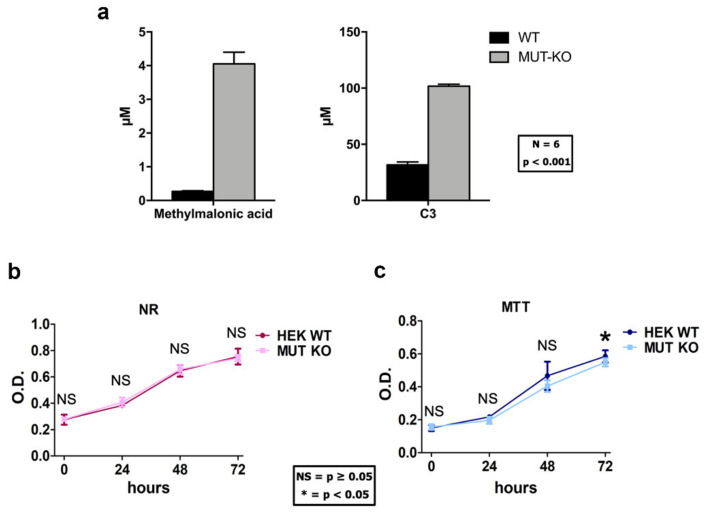
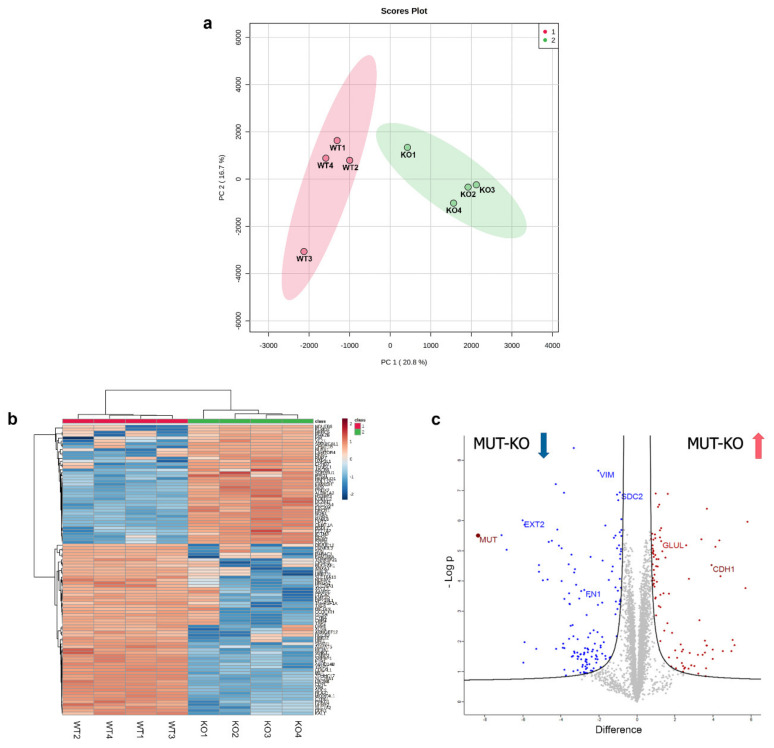
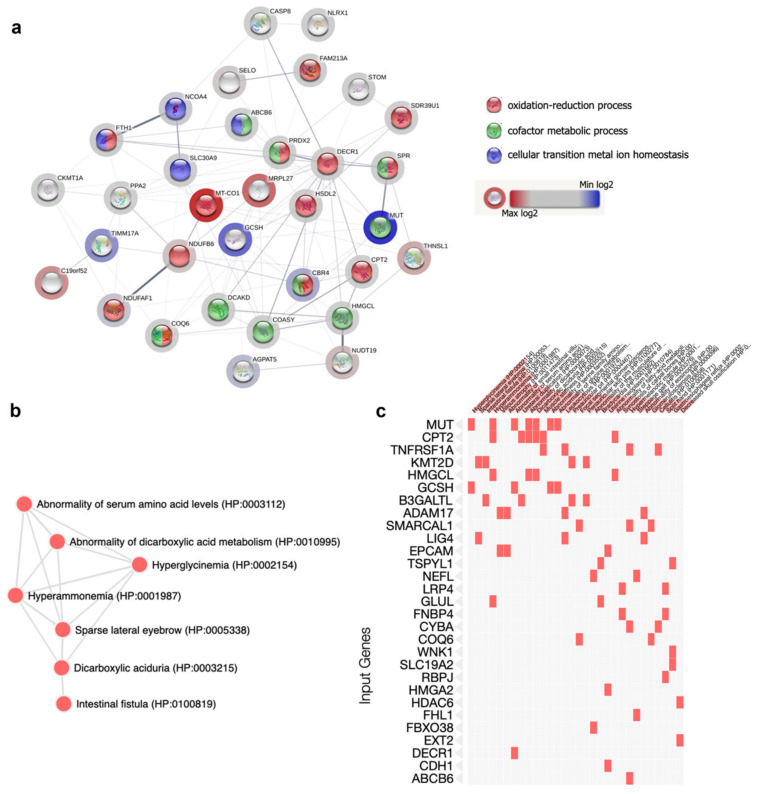
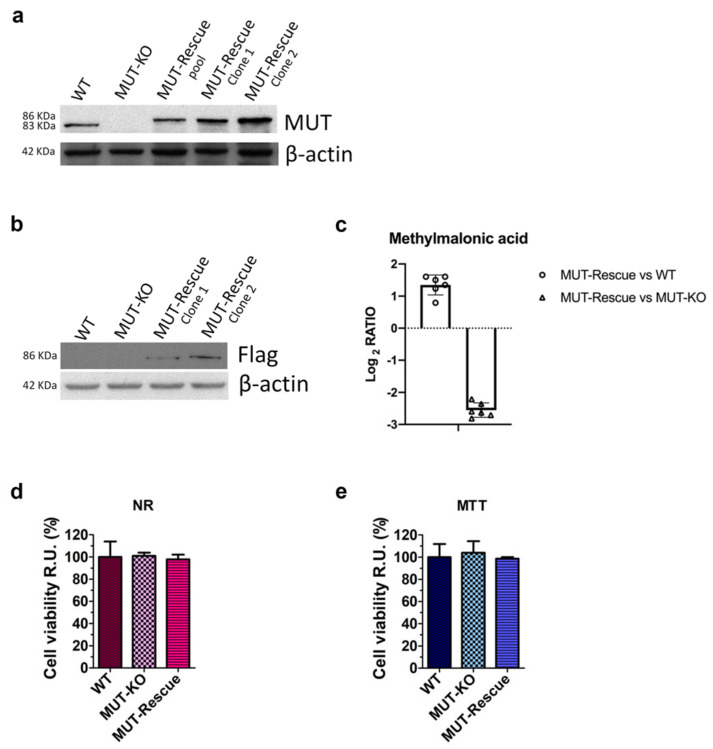
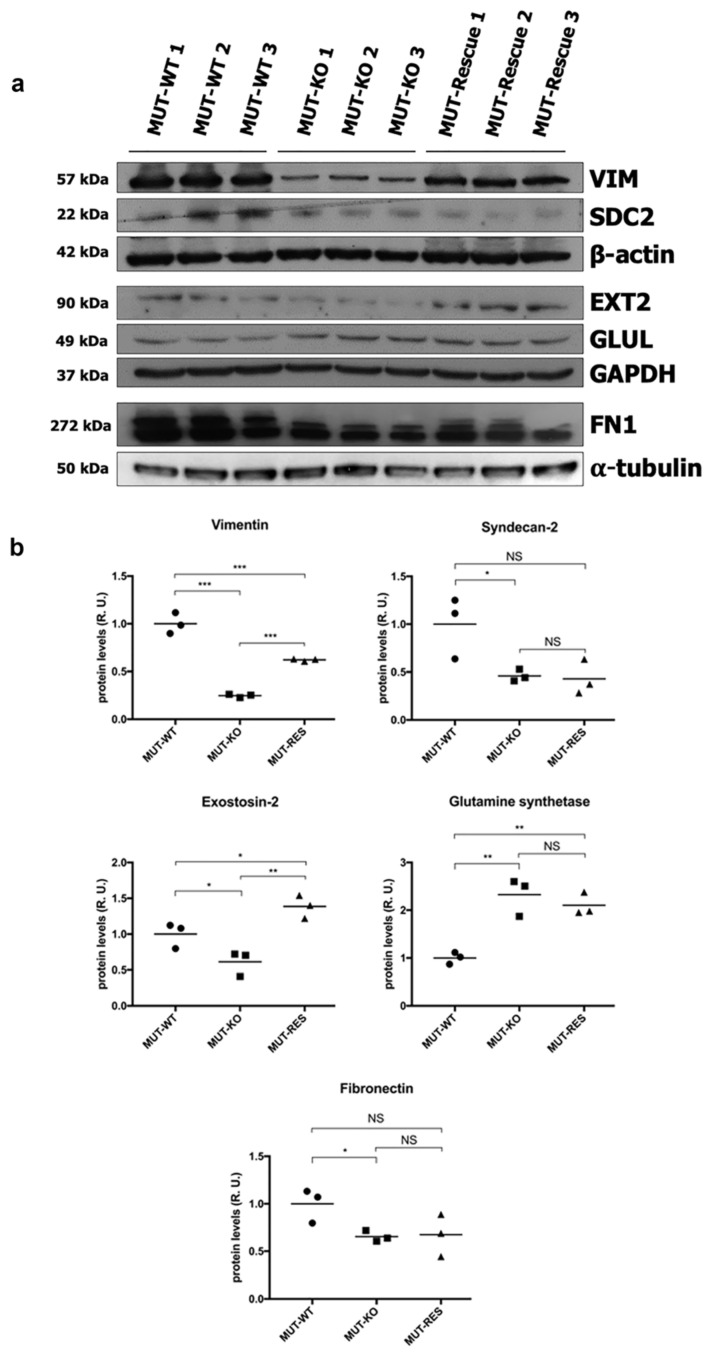
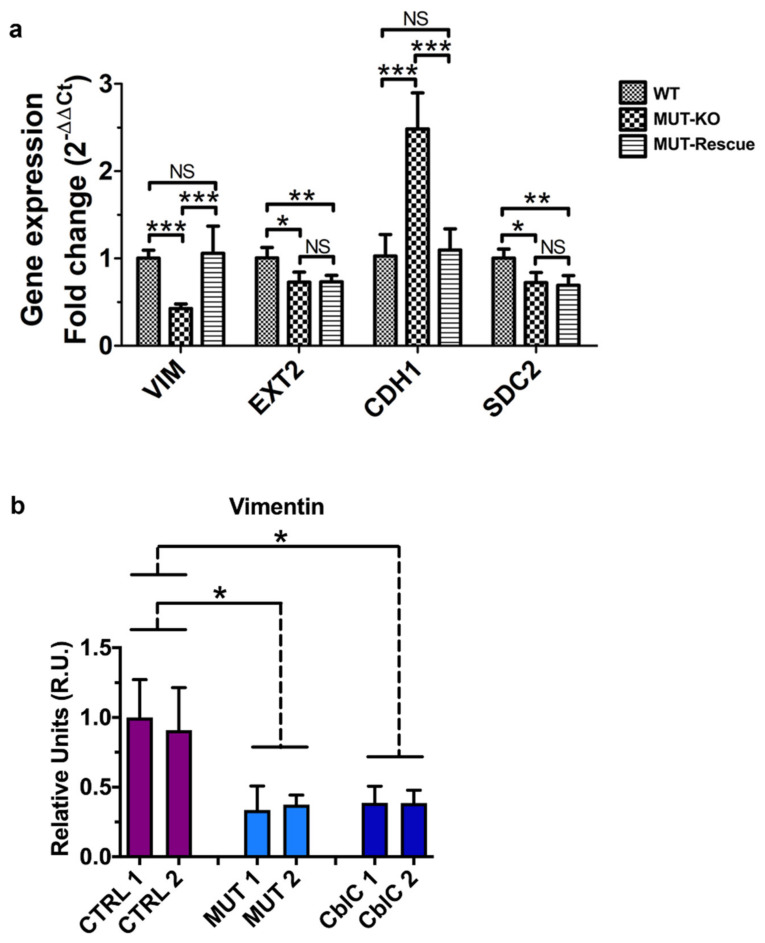
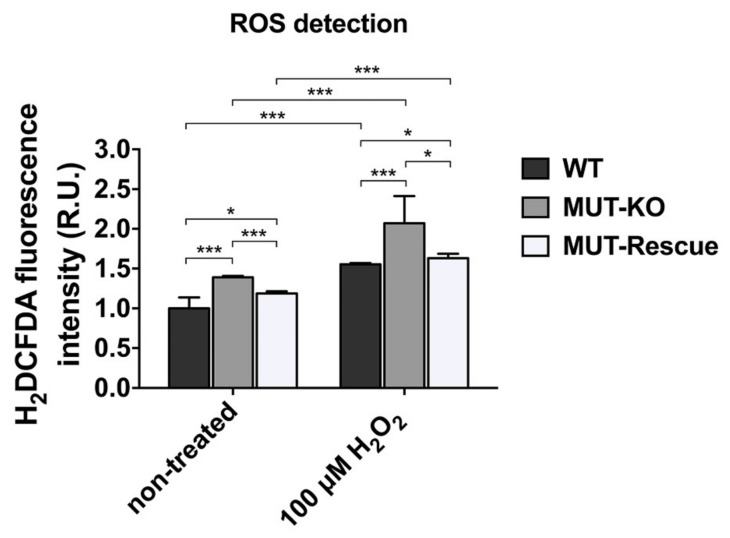
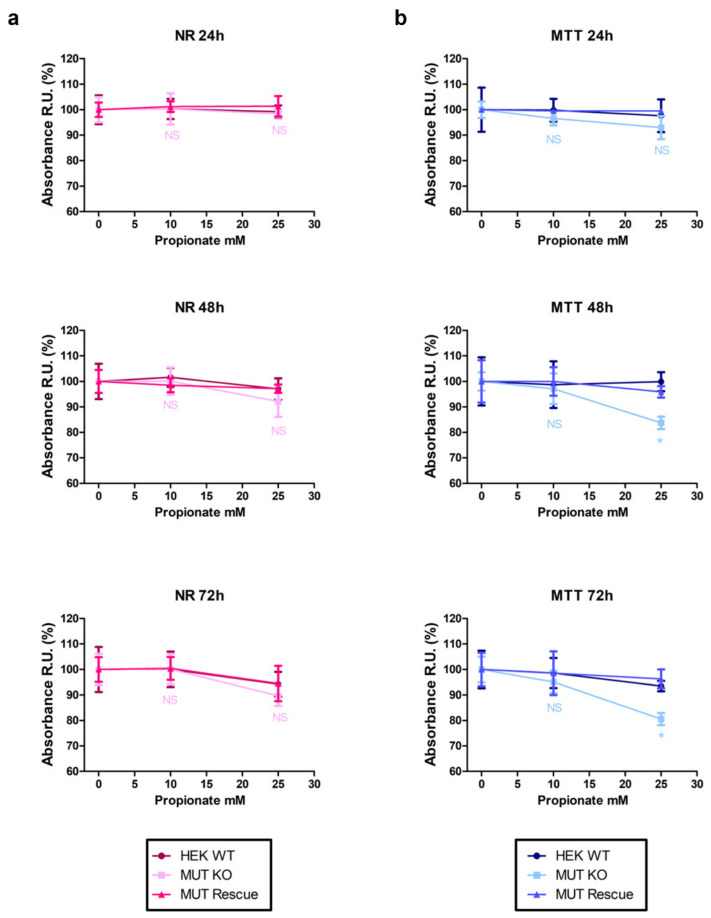
Methylmalonic acidemia (MMA) is a rare inborn error of metabolism caused by deficiency of the methylmalonyl-CoA mutase (MUT) enzyme. Downstream MUT deficiency, methylmalonic acid accumulates together with toxic metabolites from propionyl-CoA and other compounds upstream of the block in the enzyme pathway. The presentation is with life-threatening acidosis, respiratory distress, brain disturbance, hyperammonemia, and ketosis. Survivors develop poorly understood multi-organ damage, notably to the brain and kidneys. The HEK 293 cell line was engineered by CRISPR/Cas9 technology to knock out the gene (MUT-KO). Shotgun label-free quantitative proteomics and bioinformatics analyses revealed potential damaging biological processes in MUT-deficient cells. MUT-KO induced alteration of cellular architecture and morphology, and ROS overproduction. We found the alteration of proteins involved in cytoskeleton and cell adhesion organization, cell trafficking, mitochondrial, and oxidative processes, as validated by the regulation of VIM, EXT2, SDC2, FN1, GLUL, and CHD1. Additionally, a cell model of MUT-rescuing was developed in order to control the specificity of MUT-KO effects. Globally, the proteomic landscape of MUT-KO suggests the cell model to have an increased susceptibility to propionate- and HO-induced stress through an impairment of the mitochondrial functionality and unbalances in the oxidation-reduction processes.
甲基丙二酸血症(MMA)是一种罕见的先天性代谢缺陷病,由甲基丙二酰辅酶 A 变位酶(MUT)缺乏引起。下游 MUT 缺乏导致甲基丙二酸与来自丙酰辅酶 A 的毒性代谢物以及酶途径中阻断部位上游的其他化合物一起积累。临床表现为危及生命的酸中毒、呼吸窘迫、脑功能障碍、高氨血症和酮症。幸存者会出现多器官损伤,目前尚不清楚其发病机制,尤其是脑和肾脏损伤。HEK 293 细胞系通过 CRISPR/Cas9 技术进行基因敲除,构建了 MUT 基因敲除(MUT-KO)细胞系。无标记定量蛋白质组学和生物信息学分析揭示了 MUT 缺陷细胞中潜在的破坏性生物学过程。MUT-KO 诱导细胞结构和形态改变以及 ROS 过度产生。我们发现参与细胞骨架和细胞黏附组织、细胞运输、线粒体和氧化过程的蛋白质发生改变,VIM、EXT2、SDC2、FN1、GLUL 和 CHD1 的调节得到了验证。此外,还构建了 MUT 挽救的细胞模型,以控制 MUT-KO 效应的特异性。总体而言,MUT-KO 的蛋白质组学图谱表明,该细胞模型由于线粒体功能受损和氧化还原过程失衡,导致对丙酸盐和 HO 诱导的应激的敏感性增加。