Sackler Institute for Comparative Genomics, American Museum of Natural History, New York, NY, 10024, USA.
Richard Gilder Graduate School, American Museum of Natural History, New York, NY, 10024, USA.
Sci Rep. 2017 Jul 31;7(1):6589. doi: 10.1038/s41598-017-06665-3.
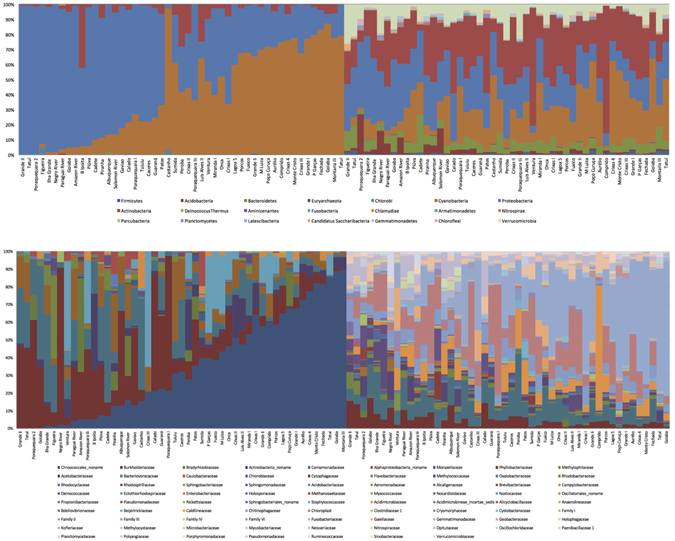
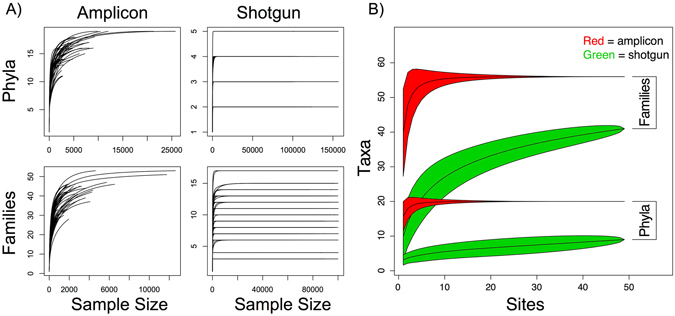
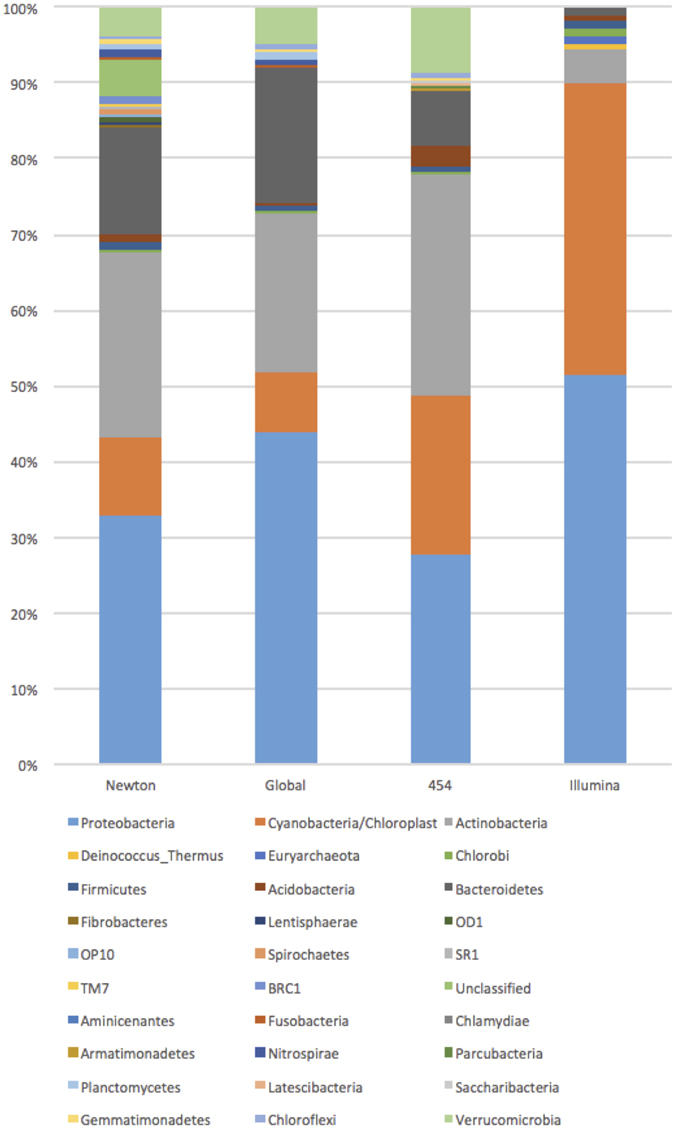
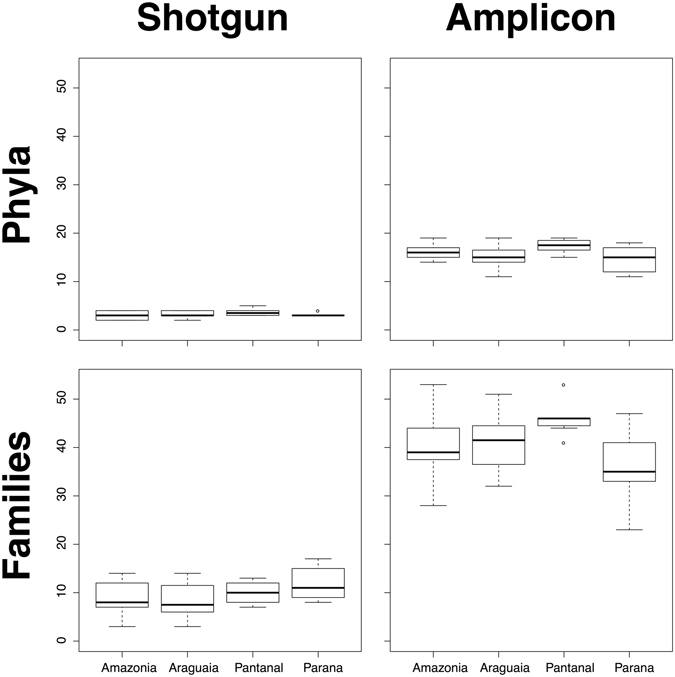
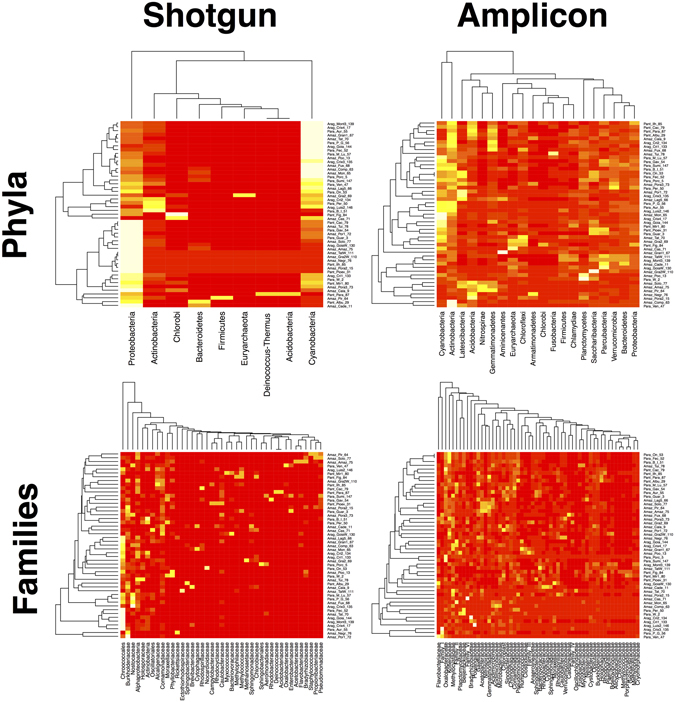
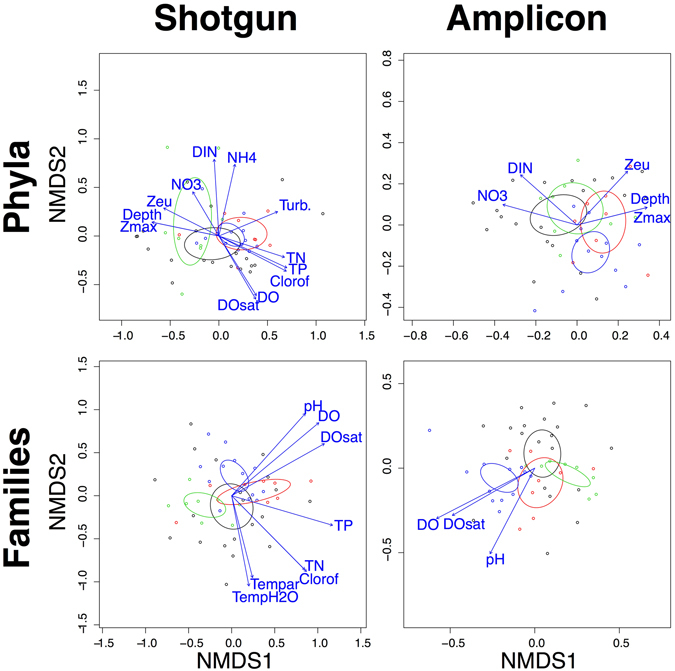
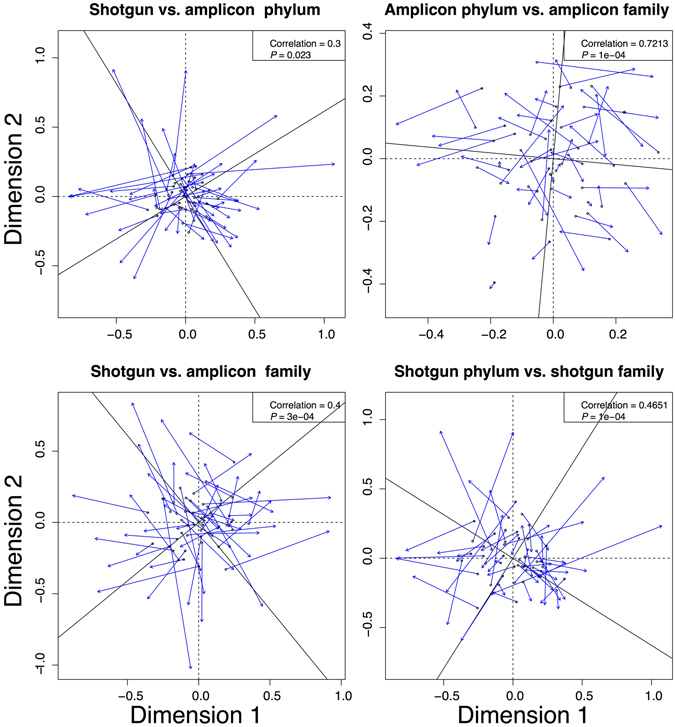
Modern metagenomic environmental DNA studies are almost completely reliant on next-generation sequencing, making evaluations of these methods critical. We compare two next-generation sequencing techniques - amplicon and shotgun - on water samples across four of Brazil's major river floodplain systems (Amazon, Araguaia, Paraná, and Pantanal). Less than 50% of phyla identified via amplicon sequencing were recovered from shotgun sequencing, clearly challenging the dogma that mid-depth shotgun recovers more diversity than amplicon-based approaches. Amplicon sequencing also revealed ~27% more families. Overall the amplicon data were more robust across both biodiversity and community ecology analyses at different taxonomic scales. Our work doubles the sampling size in similar environmental studies, and novelly integrates environmental data (e.g., pH, temperature, nutrients) from each site, revealing divergent correlations depending on which data are used. While myriad variants on NGS techniques and bioinformatic pipelines are available, our results point to core differences that have not been highlighted in any studies to date. Given the low number of taxa identified when coupling shotgun data with clade-based taxonomic algorithms, previous studies that quantified biodiversity using such bioinformatic tools should be viewed cautiously or re-analyzed. Nonetheless, shotgun has complementary advantages that should be weighed when designing projects.
现代宏基因组环境 DNA 研究几乎完全依赖于下一代测序技术,因此对这些方法进行评估至关重要。我们比较了两种下一代测序技术——扩增子和鸟枪法——在巴西四个主要河流泛滥平原系统(亚马逊、阿拉瓜亚、巴拉那和潘塔纳尔)的水样中的应用。从鸟枪法中回收的门的数量不到扩增子测序的 50%,这显然对深度鸟枪法比基于扩增子的方法能回收更多多样性的教条提出了挑战。扩增子测序还揭示了约 27%更多的科。总的来说,在不同的分类尺度上,扩增子数据在生物多样性和群落生态学分析方面都更稳健。我们的工作将类似环境研究中的采样规模扩大了一倍,并创新性地整合了每个地点的环境数据(如 pH 值、温度、养分),根据所使用的数据揭示了不同的相关性。虽然有大量的 NGS 技术和生物信息学管道变体可供选择,但我们的结果指出了迄今为止任何研究都没有强调的核心差异。鉴于与基于进化枝的分类算法结合使用鸟枪法数据时识别出的分类群数量较少,使用此类生物信息学工具量化生物多样性的先前研究应谨慎看待或重新分析。尽管如此,鸟枪法具有互补的优势,在设计项目时应权衡这些优势。