Dept. of Plant Sciences and Center for Population Biology, University of California, Davis, Davis, CA, USA.
Dept. of Ecology and Evolutionary Biology, University of California, Irvine, Irvine, CA, USA.
PLoS Genet. 2018 Nov 19;14(11):e1007794. doi: 10.1371/journal.pgen.1007794. eCollection 2018 Nov.
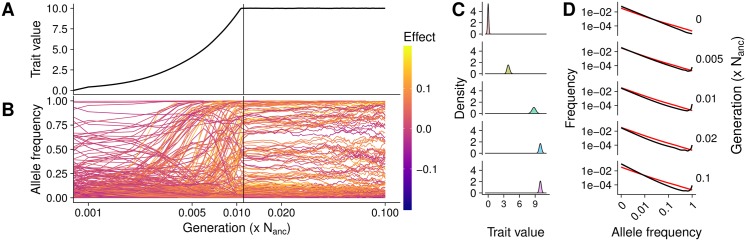
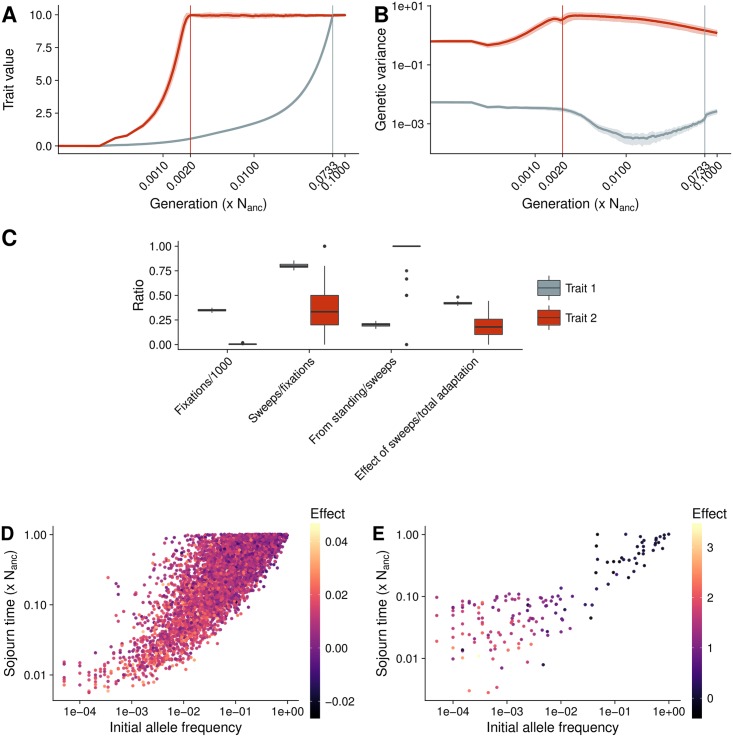
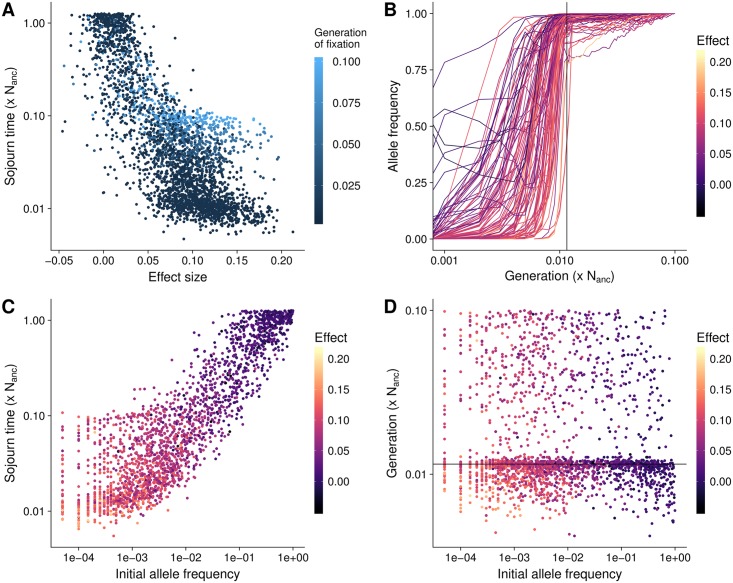
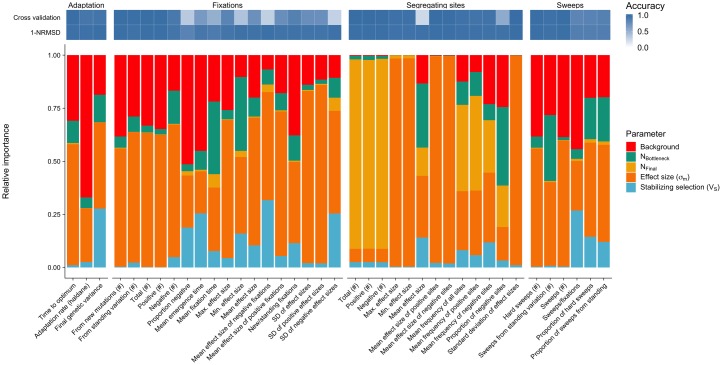
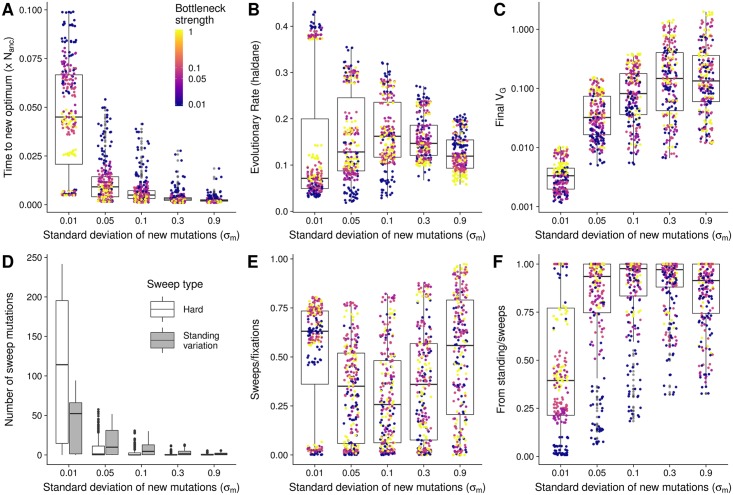
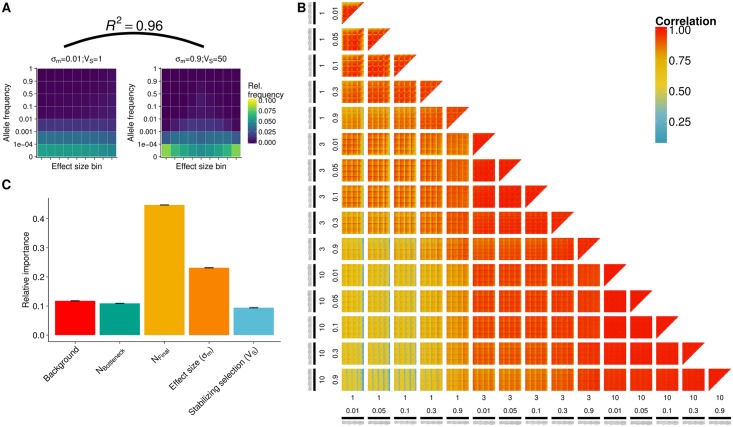
Understanding the genetic basis of phenotypic adaptation to changing environments is an essential goal of population and quantitative genetics. While technological advances now allow interrogation of genome-wide genotyping data in large panels, our understanding of the process of polygenic adaptation is still limited. To address this limitation, we use extensive forward-time simulation to explore the impacts of variation in demography, trait genetics, and selection on the rate and mode of adaptation and the resulting genetic architecture. We simulate a population adapting to an optimum shift, modeling sequence variation for 20 QTL for each of 12 different demographies for 100 different traits varying in the effect size distribution of new mutations, the strength of stabilizing selection, and the contribution of the genomic background. We then use random forest regression approaches to learn the relative importance of input parameters in determining a number of aspects of the process of adaptation, including the speed of adaptation, the relative frequency of hard sweeps and sweeps from standing variation, or the final genetic architecture of the trait. We find that selective sweeps occur even for traits under relatively weak selection and where the genetic background explains most of the variation. Though most sweeps occur from variation segregating in the ancestral population, new mutations can be important for traits under strong stabilizing selection that undergo a large optimum shift. We also show that population bottlenecks and expansion impact overall genetic variation as well as the relative importance of sweeps from standing variation and the speed with which adaptation can occur. We then compare our results to two traits under selection during maize domestication, showing that our simulations qualitatively recapitulate differences between them. Overall, our results underscore the complex population genetics of individual loci in even relatively simple quantitative trait models, but provide a glimpse into the factors that drive this complexity and the potential of these approaches for understanding polygenic adaptation.
理解表型适应不断变化环境的遗传基础是群体和数量遗传学的一个重要目标。虽然技术进步现在允许在大型面板中检查全基因组基因分型数据,但我们对多基因适应过程的理解仍然有限。为了解决这一限制,我们使用广泛的正向时间模拟来探索人口统计学、性状遗传学和选择的变化对适应速度和模式以及由此产生的遗传结构的影响。我们模拟了一个适应最佳转移的种群,为 12 种不同人口统计学中每一种的 100 个不同性状的 20 个 QTL 建模序列变异,这些性状的新突变效应大小分布、稳定选择的强度和基因组背景的贡献不同。然后,我们使用随机森林回归方法来学习输入参数在确定适应过程的许多方面的相对重要性,包括适应速度、硬选择和来自固定变异的选择的相对频率,或性状的最终遗传结构。我们发现,即使在相对较弱的选择下,而且遗传背景解释了大部分变异的情况下,选择也会发生。虽然大多数选择发生在祖先群体中分离的变异中,但对于经历较大最佳转移的受强烈稳定选择的性状,新突变可能很重要。我们还表明,种群瓶颈和扩张会影响整体遗传变异,以及来自固定变异的选择和适应发生速度的相对重要性。然后,我们将我们的结果与玉米驯化过程中选择的两个性状进行比较,表明我们的模拟定性地再现了它们之间的差异。总的来说,我们的结果强调了即使在相对简单的数量性状模型中,单个基因座的复杂群体遗传学,但提供了一个了解驱动这种复杂性的因素以及这些方法用于理解多基因适应的潜力的视角。