Department of Genetics, Microbiology and Statistics, Faculty of Biology, Universitat de Barcelona, Diagonal, 643, 08028, Barcelona, Catalonia, Spain.
Department of Osteopathic Medical Specialties, Michigan State University, 909 Fee Road, Room B 309 West Fee Hall, East Lansing, MI, 48824, USA.
BMC Bioinformatics. 2018 Nov 19;19(1):432. doi: 10.1186/s12859-018-2451-4.
Support vector machines (SVM) are a powerful tool to analyze data with a number of predictors approximately equal or larger than the number of observations. However, originally, application of SVM to analyze biomedical data was limited because SVM was not designed to evaluate importance of predictor variables. Creating predictor models based on only the most relevant variables is essential in biomedical research. Currently, substantial work has been done to allow assessment of variable importance in SVM models but this work has focused on SVM implemented with linear kernels. The power of SVM as a prediction model is associated with the flexibility generated by use of non-linear kernels. Moreover, SVM has been extended to model survival outcomes. This paper extends the Recursive Feature Elimination (RFE) algorithm by proposing three approaches to rank variables based on non-linear SVM and SVM for survival analysis.
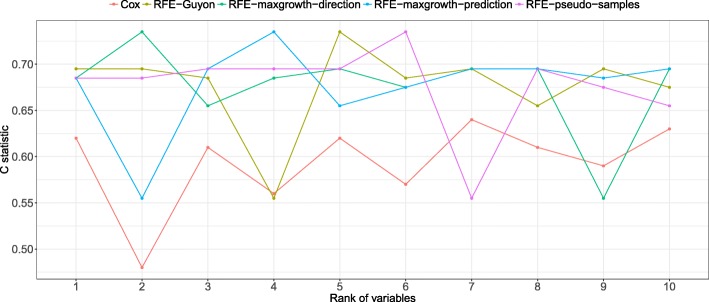
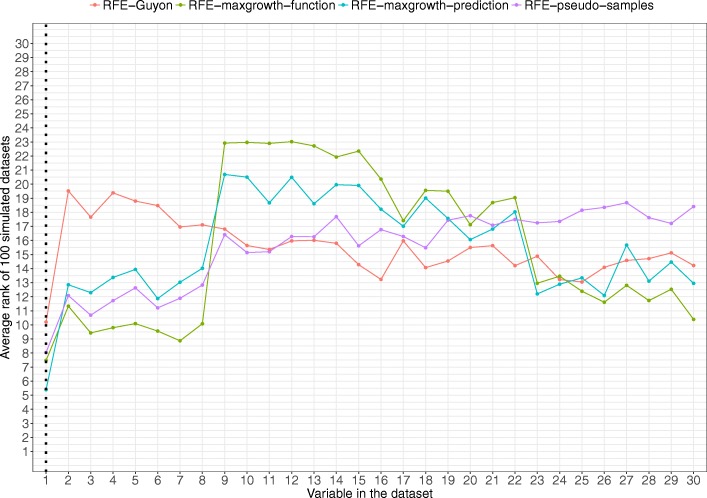
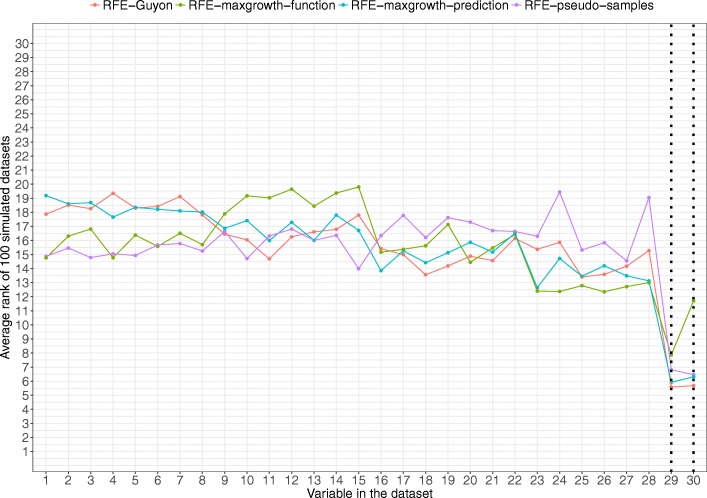
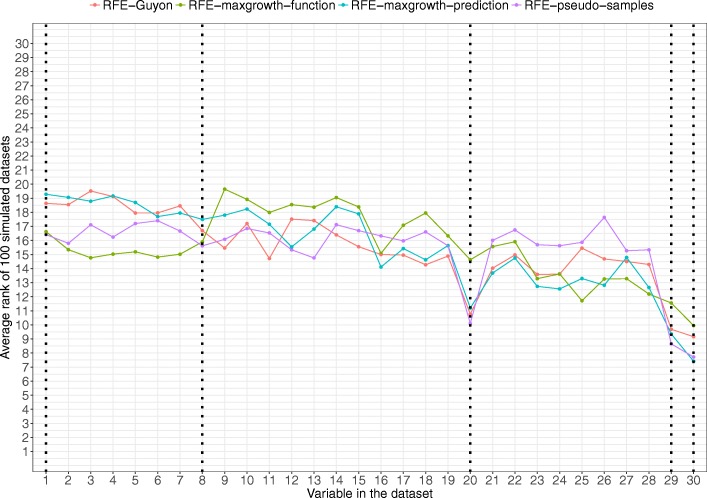
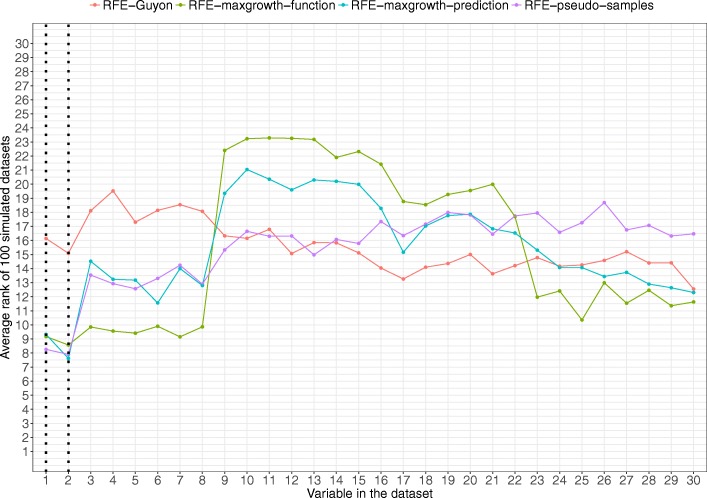
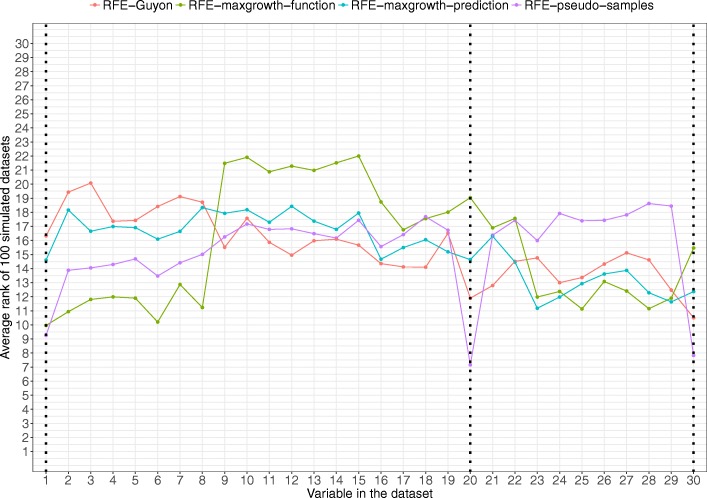
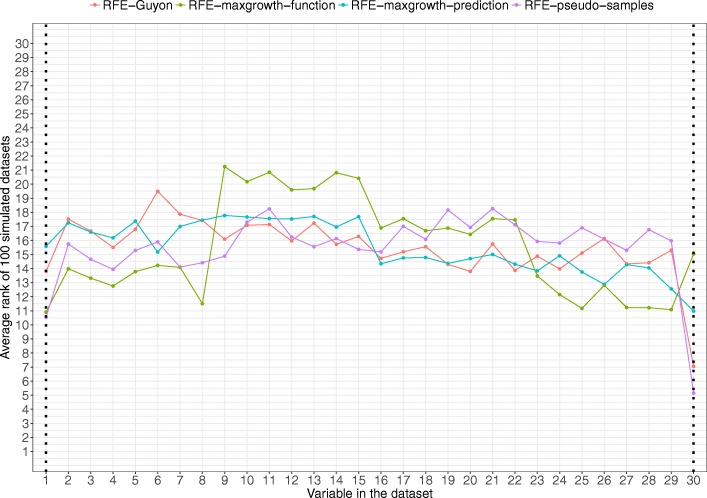
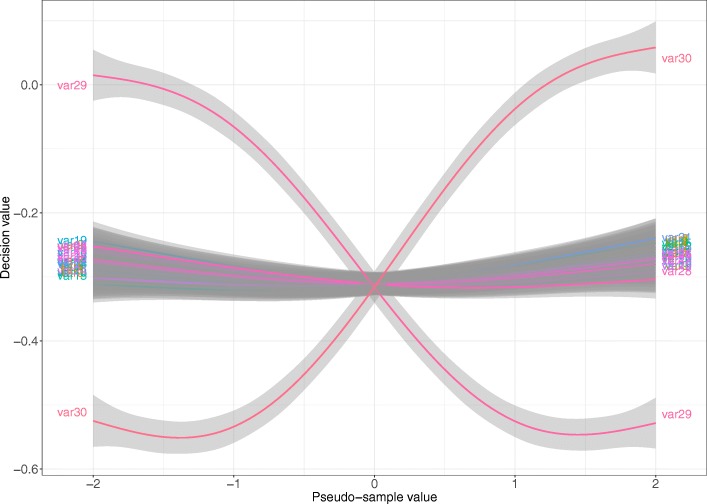
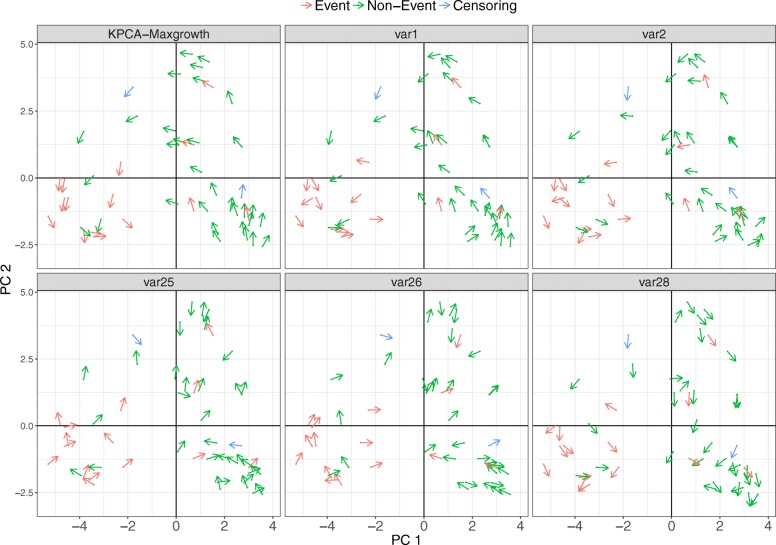
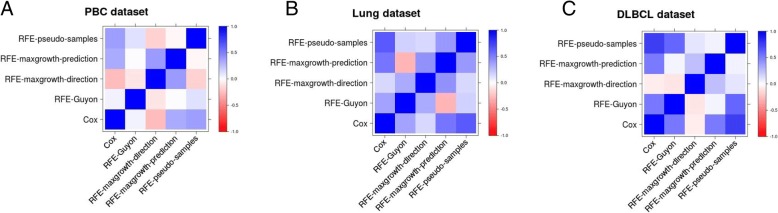
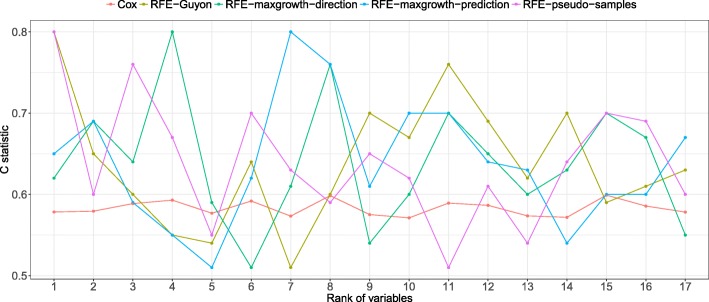
The proposed algorithms allows visualization of each one the RFE iterations, and hence, identification of the most relevant predictors of the response variable. Using simulation studies based on time-to-event outcomes and three real datasets, we evaluate the three methods, based on pseudo-samples and kernel principal component analysis, and compare them with the original SVM-RFE algorithm for non-linear kernels. The three algorithms we proposed performed generally better than the gold standard RFE for non-linear kernels, when comparing the truly most relevant variables with the variable ranks produced by each algorithm in simulation studies. Generally, the RFE-pseudo-samples outperformed the other three methods, even when variables were assumed to be correlated in all tested scenarios.
The proposed approaches can be implemented with accuracy to select variables and assess direction and strength of associations in analysis of biomedical data using SVM for categorical or time-to-event responses. Conducting variable selection and interpreting direction and strength of associations between predictors and outcomes with the proposed approaches, particularly with the RFE-pseudo-samples approach can be implemented with accuracy when analyzing biomedical data. These approaches, perform better than the classical RFE of Guyon for realistic scenarios about the structure of biomedical data.
支持向量机(SVM)是一种强大的工具,可用于分析具有与观测值数量大致相等或更多预测变量的数据。然而,最初,SVM 应用于生物医学数据分析受到限制,因为 SVM 不是为评估预测变量的重要性而设计的。在生物医学研究中,基于最相关的变量创建预测模型是至关重要的。目前,已经做了大量工作来允许在 SVM 模型中评估变量的重要性,但这项工作主要集中在具有线性核的 SVM 上。SVM 作为预测模型的强大功能与使用非线性核产生的灵活性有关。此外,SVM 已扩展到用于建模生存结果。本文通过提出三种基于非线性 SVM 和 SVM 进行生存分析的变量排名方法,扩展了递归特征消除(RFE)算法。
所提出的算法允许可视化每个 RFE 迭代,因此,可以识别出与响应变量最相关的预测变量。我们使用基于时间到事件结果的模拟研究和三个真实数据集,基于伪样本和核主成分分析评估了这三种方法,并将它们与用于非线性核的原始 SVM-RFE 算法进行了比较。在所提出的三种算法中,在比较模拟研究中真正最相关的变量与每个算法生成的变量等级时,与非线性核的黄金标准 RFE 相比,一般表现得更好。通常,RFE-伪样本优于其他三种方法,即使在所有测试场景中都假设变量是相关的。
在所提出的方法中,可以使用准确性来选择变量,并使用 SVM 分析生物医学数据中的分类或时间到事件响应,评估预测变量和响应之间的关联方向和强度。在分析生物医学数据时,特别是使用 RFE-伪样本方法,这些方法可以准确地执行变量选择并解释预测变量和结果之间的关联方向和强度。与 Guyon 的经典 RFE 相比,这些方法在关于生物医学数据结构的现实场景中表现更好。