Department of Molecular, Cellular and Developmental Biology, University of Colorado Boulder, Boulder, CO, USA.
Inscripta, Inc., Boulder, CO, USA.
Mol Syst Biol. 2018 Nov 26;14(11):e8371. doi: 10.15252/msb.20188371.
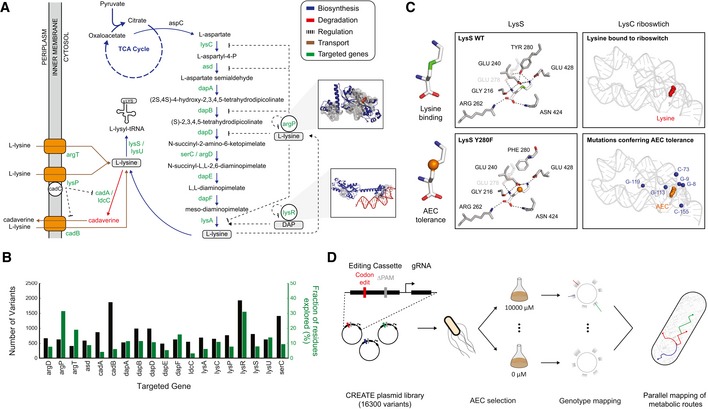
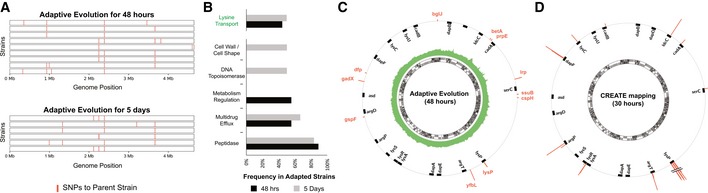
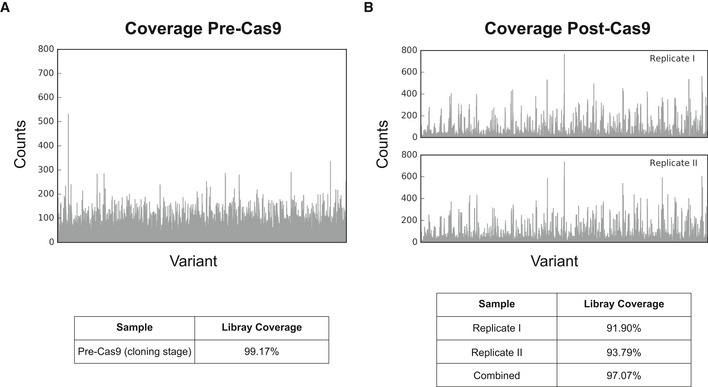
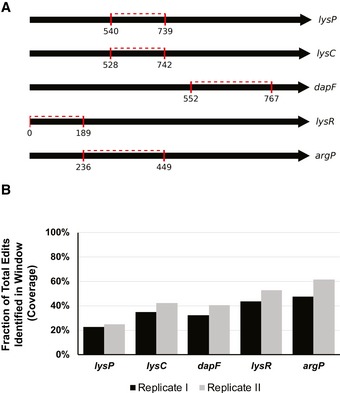
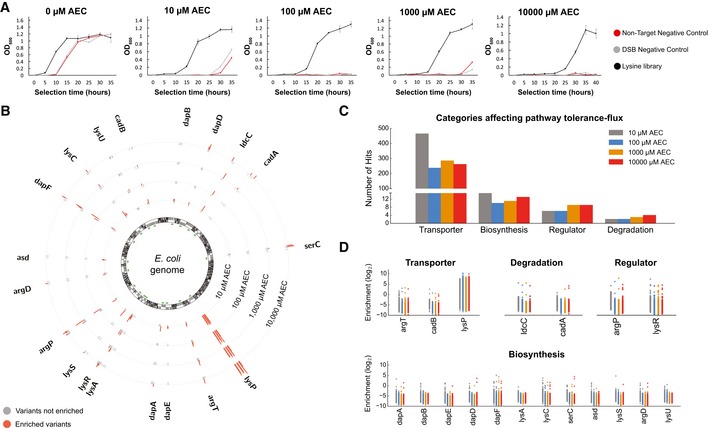
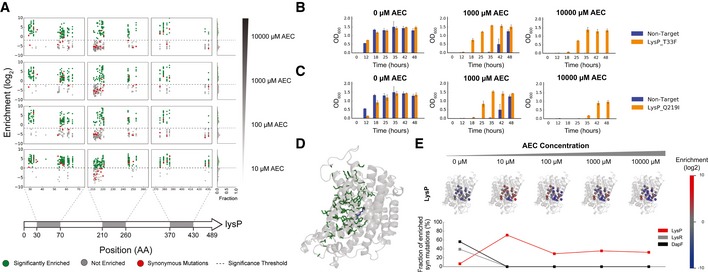
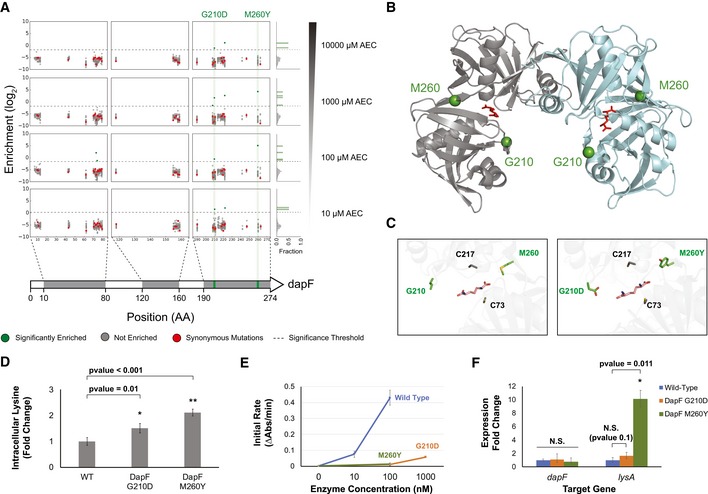
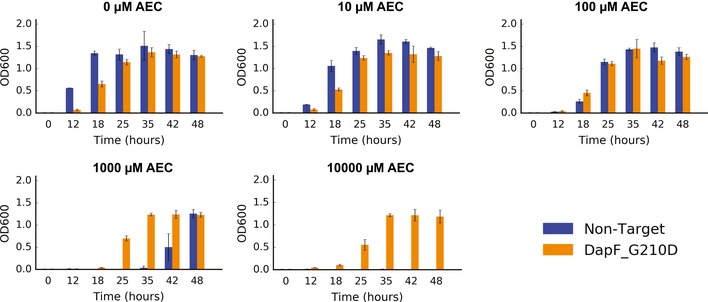
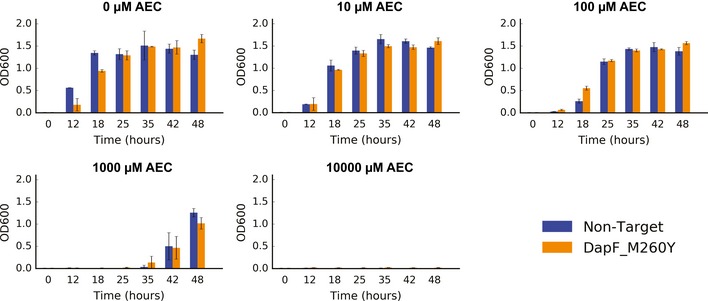
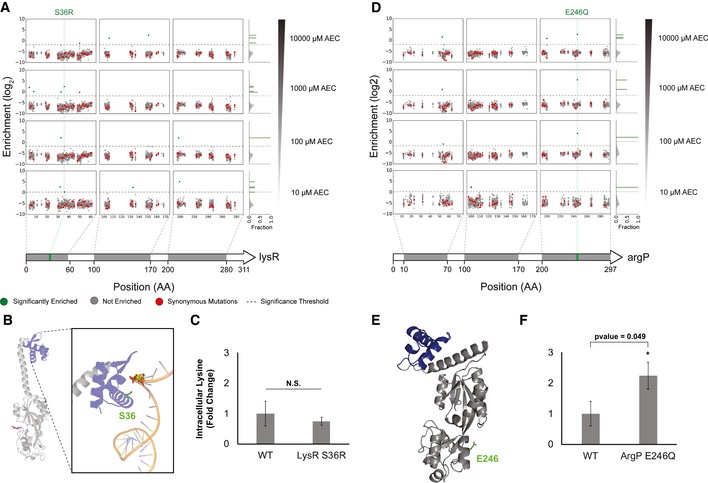
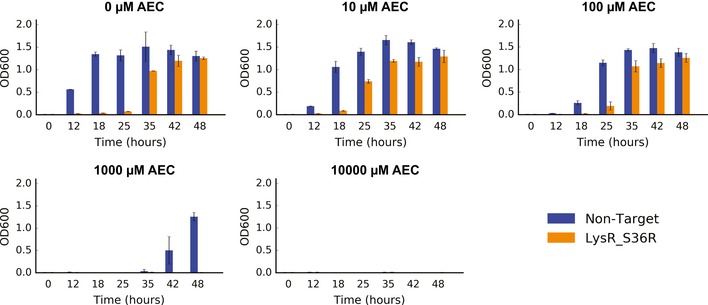
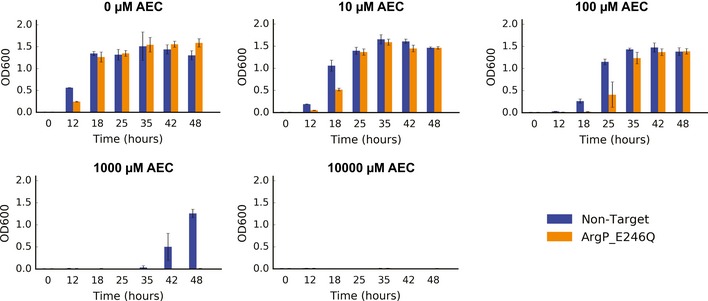
Our limited ability to predict genotype-phenotype relationships has called for strategies that allow testing of thousands of hypotheses in parallel. Deep scanning mutagenesis has been successfully implemented to map genotype-phenotype relationships at a single-protein scale, allowing scientists to elucidate properties that are difficult to predict. However, most phenotypes are dictated by several proteins that are interconnected through complex and robust regulatory and metabolic networks. These sophisticated networks hinder our understanding of the phenotype of interest and limit our capabilities to rewire cellular functions. Here, we leveraged CRISPR-EnAbled Trackable genome Engineering to attempt a parallel and high-resolution interrogation of complex networks, deep scanning multiple proteins associated with lysine metabolism in We designed over 16,000 mutations to perturb this pathway and mapped their contribution toward resistance to an amino acid analog. By doing so, we identified different routes that can alter pathway function and flux, uncovering mechanisms that would be difficult to rationally design. This approach sets a framework for forward investigation of complex multigenic phenotypes.
我们预测基因型-表型关系的能力有限,这就需要采用能够并行测试数千个假设的策略。深度扫描诱变已成功用于在单个蛋白质尺度上绘制基因型-表型关系,使科学家能够阐明难以预测的特性。然而,大多数表型是由几种通过复杂而稳健的调节和代谢网络相互连接的蛋白质决定的。这些复杂的网络阻碍了我们对感兴趣的表型的理解,并限制了我们重新布线细胞功能的能力。在这里,我们利用 CRISPR-EnAbled Trackable genome Engineering 尝试对复杂网络进行并行和高分辨率的探究,深度扫描与赖氨酸代谢相关的多种蛋白质。我们设计了超过 16000 个突变来扰乱这条途径,并绘制了它们对氨基酸类似物抗性的贡献。通过这样做,我们确定了可以改变途径功能和通量的不同途径,揭示了难以通过理性设计来揭示的机制。这种方法为复杂多基因表型的前瞻性研究奠定了框架。