Department of Animal Medicine, Production and Health, University of Padova, Legnaro, Italy.
UAB, Centre de Recerca en Sanitat Animal (CRESA, IRTA-UAB), Campus de la Universitat Autònoma de Barcelona, Bellaterra, Spain.
PLoS One. 2018 Dec 6;13(12):e0208585. doi: 10.1371/journal.pone.0208585. eCollection 2018.
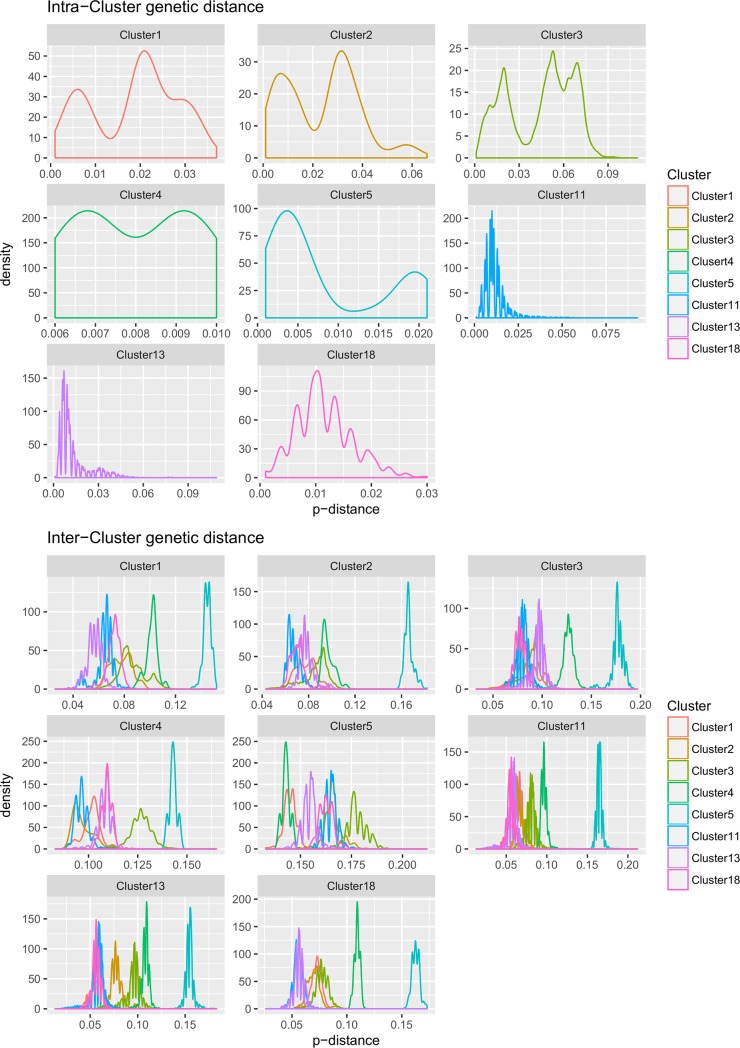
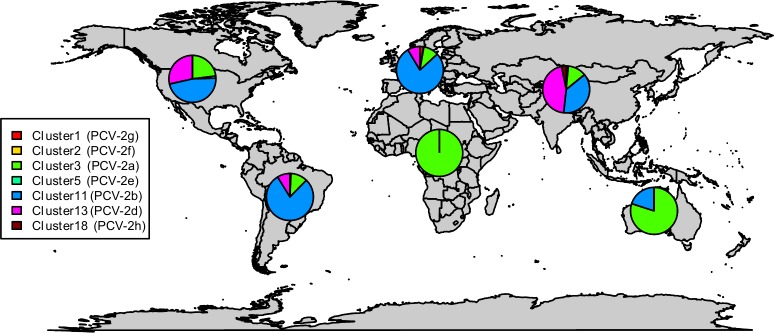
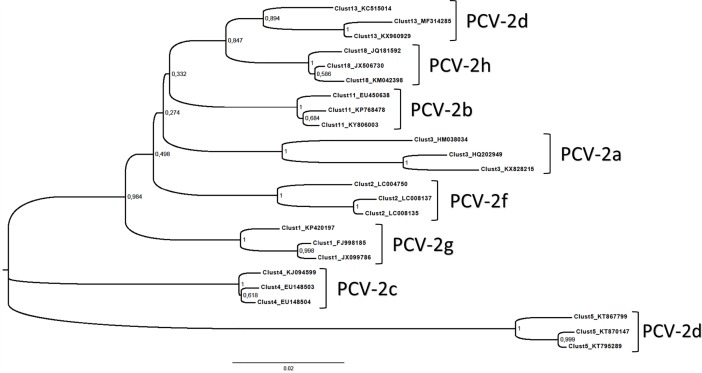
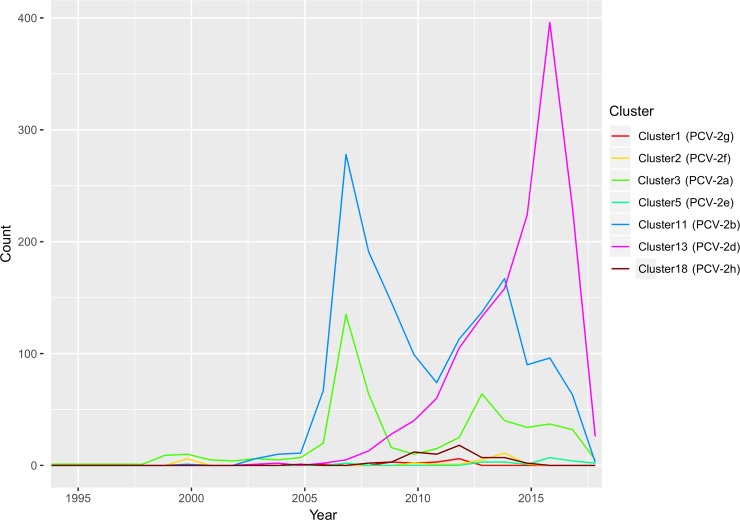
Porcine circovirus 2 (PCV-2) is one of the most widespread viral infections of swine, causing a remarkable economic impact because of direct losses and indirect costs for its control. As other ssDNA viruses, PCV-2 is characterized by a high evolutionary rate, leading to the emergence of a plethora of variants with different biological and epidemiological features. Over time, several attempts have been made to organize PCV-2 genetic heterogeneity in recognized genotypes. This categorization has clearly simplified the epidemiological investigations, allowing to identify different spatial and temporal patterns among genotypes. Additionally, variable virulence and vaccine effectiveness have also been hypothesized. However, the rapid increase in sequencing activity, coupled with the per se high viral variability, has challenged the previously established nomenclature, leading to the definition of several study-specific genotypes and hindering the capability of performing comparable epidemiological studies. Based on these premises, an updated classification scheme is herein reported. Recognizing the impossibility of defining a clear inter-cluster p-distance cut-off, the present study proposes a phylogeny-grounded genotype definition based on three criteria: maximum intra-genotype p-distance of 13% (calculated on the ORF2 gene), bootstrap support at the corresponding internal node higher than 70% and at least 15 available sequences. This scheme allowed defining 8 genotypes (PCV-2a to PCV-2h), which six of those had been previously proposed. To minimize the inconvenience of implementing a new classification, the most common names already adopted have been maintained when possible. The analysis of sequence-associated metadata highlighted a highly unbalanced sequencing activity in terms of geographical, host and temporal distribution. The PCV-2 molecular epidemiology scenario appears therefore characterized by a severe bias that could lead to spurious associations between genetic and epidemiological/biological viral features. While the suggested classification can establish a "common language" for future studies, further efforts should be paid to achieve a more homogeneous and informative representation of the PCV-2 global scenario.
猪圆环病毒 2 型(PCV-2)是猪最广泛传播的病毒感染之一,因其直接损失和控制成本的间接成本而造成显著的经济影响。与其他 ssDNA 病毒一样,PCV-2 的进化速度很高,导致出现了大量具有不同生物学和流行病学特征的变体。随着时间的推移,人们已经尝试了多种方法来组织 PCV-2 的遗传异质性,以识别不同的基因型。这种分类方法显然简化了流行病学研究,能够识别不同基因型之间的空间和时间模式。此外,还假设了不同的毒力和疫苗效果。然而,测序活动的快速增加,加上病毒本身的高度可变性,对先前建立的命名法提出了挑战,导致定义了几个特定于研究的基因型,并阻碍了进行可比的流行病学研究的能力。基于这些前提,本文报告了一个更新的分类方案。鉴于无法定义明确的聚类间 p-距离截止值,本研究提出了一种基于三个标准的基于系统发育的基因型定义:ORF2 基因上计算的最大基因型内 p-距离为 13%,相应内部节点的自举支持率高于 70%,至少有 15 个可用序列。该方案定义了 8 个基因型(PCV-2a 至 PCV-2h),其中 6 个已被提议。为了尽量减少实施新分类的不便,只要有可能,就保留了已经采用的最常见名称。与序列相关的元数据的分析突出了地理、宿主和时间分布方面高度不平衡的测序活动。因此,PCV-2 的分子流行病学情况的特点是存在严重的偏差,这可能导致遗传和流行病学/生物学病毒特征之间产生虚假关联。虽然建议的分类可以为未来的研究建立“共同语言”,但仍需要进一步努力,以实现对 PCV-2 全球情况更具代表性和更具信息性的表示。