Department of Population Health, and Reproduction, School of Veterinary Medicine, University of California Davis, One Shields Ave, Davis, CA 95616, USA.
Molecular, Cellular, and Integrative Physiology Graduate Group, University of California Davis, One Shields Ave, Davis, CA 95616, USA.
Gigascience. 2019 Apr 1;8(4). doi: 10.1093/gigascience/giy158.
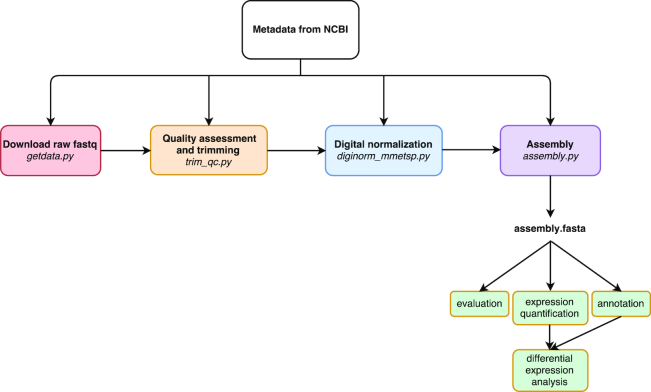
De novo transcriptome assemblies are required prior to analyzing RNA sequencing data from a species without an existing reference genome or transcriptome. Despite the prevalence of transcriptomic studies, the effects of using different workflows, or "pipelines," on the resulting assemblies are poorly understood. Here, a pipeline was programmatically automated and used to assemble and annotate raw transcriptomic short-read data collected as part of the Marine Microbial Eukaryotic Transcriptome Sequencing Project. The resulting transcriptome assemblies were evaluated and compared against assemblies that were previously generated with a different pipeline developed by the National Center for Genome Research.
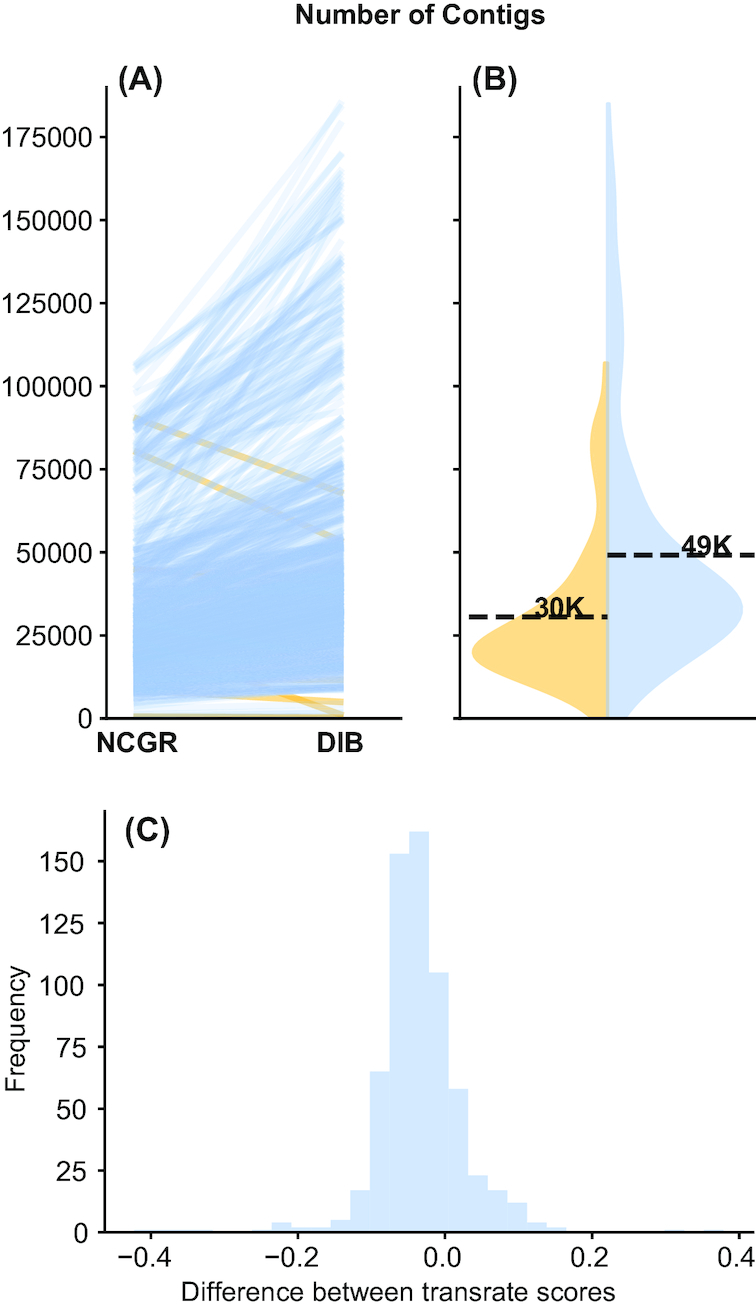
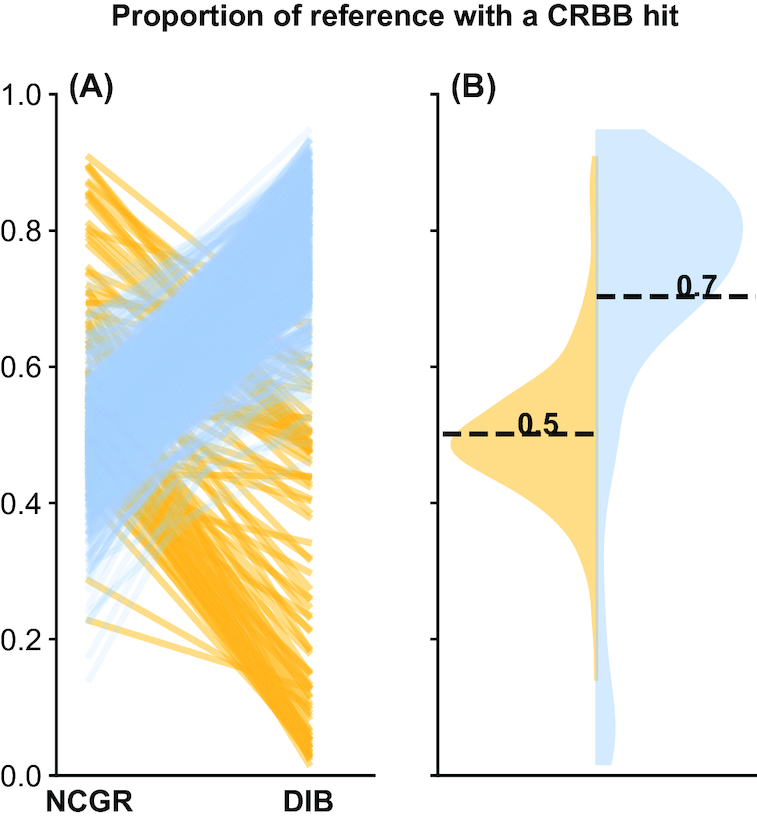
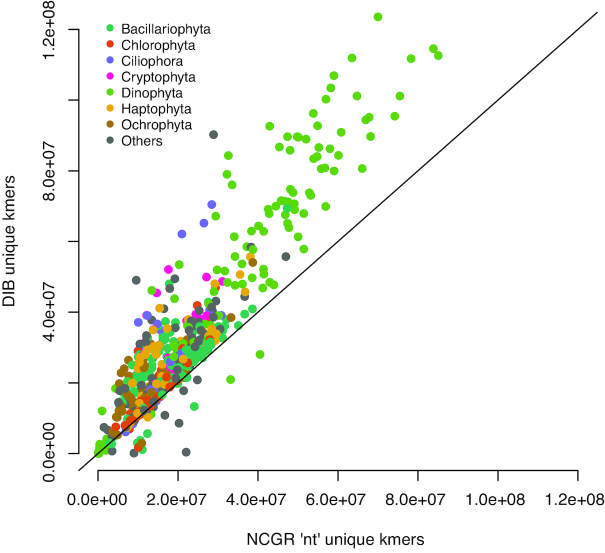
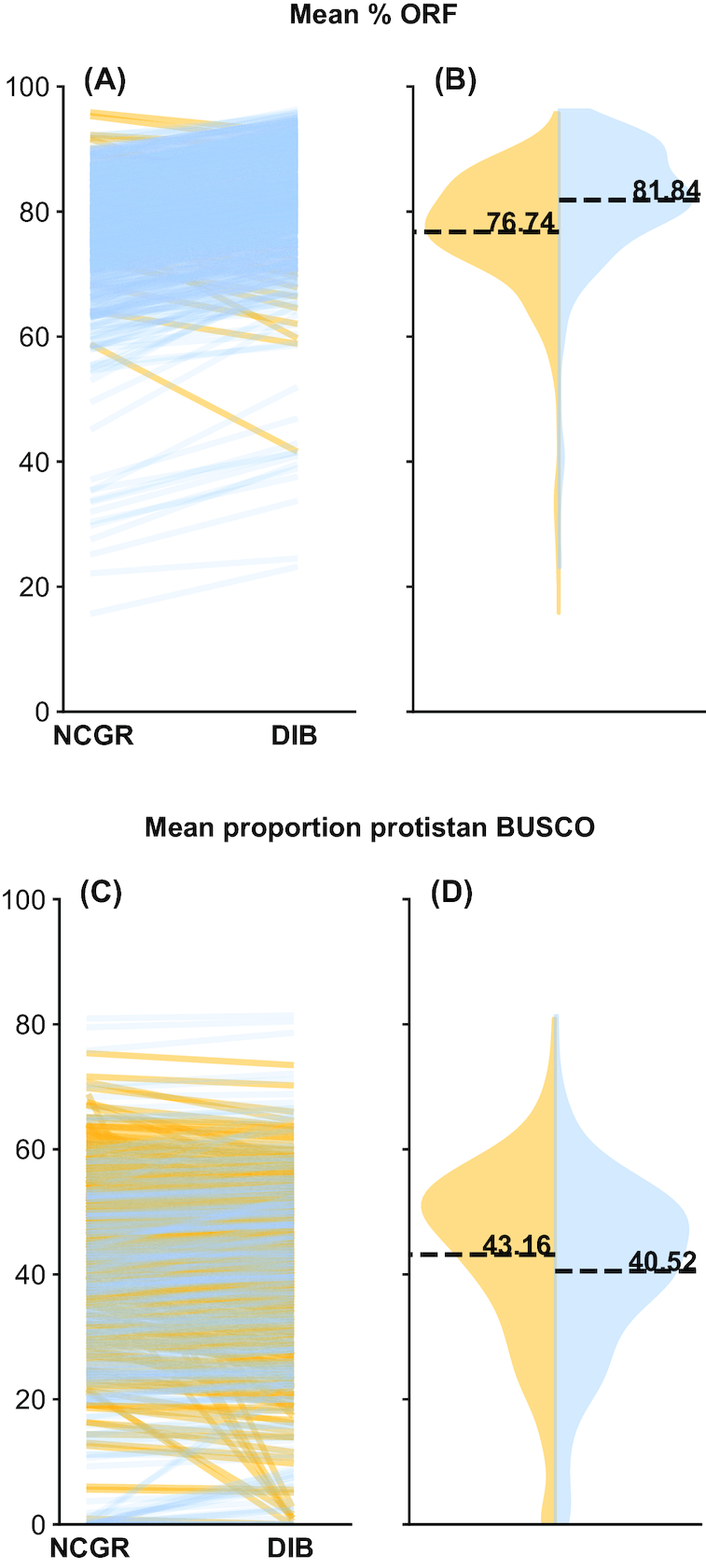
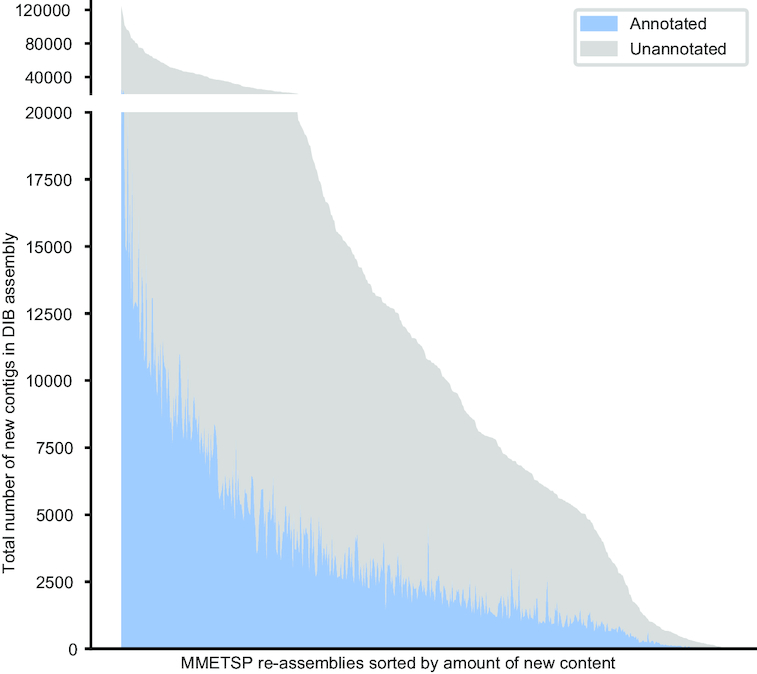
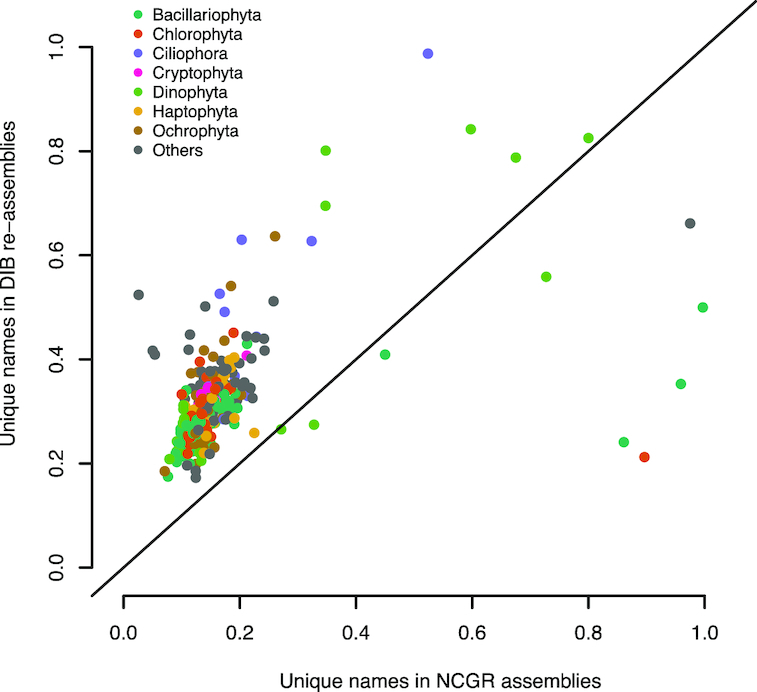
New transcriptome assemblies contained the majority of previous contigs as well as new content. On average, 7.8% of the annotated contigs in the new assemblies were novel gene names not found in the previous assemblies. Taxonomic trends were observed in the assembly metrics. Assemblies from the Dinoflagellata showed a higher number of contigs and unique k-mers than transcriptomes from other phyla, while assemblies from Ciliophora had a lower percentage of open reading frames compared to other phyla.
Given current bioinformatics approaches, there is no single "best" reference transcriptome for a particular set of raw data. As the optimum transcriptome is a moving target, improving (or not) with new tools and approaches, automated and programmable pipelines are invaluable for managing the computationally intensive tasks required for re-processing large sets of samples with revised pipelines and ensuring a common evaluation workflow is applied to all samples. Thus, re-assembling existing data with new tools using automated and programmable pipelines may yield more accurate identification of taxon-specific trends across samples in addition to novel and useful products for the community.
在分析没有现有参考基因组或转录组的物种的 RNA 测序数据之前,需要从头转录组组装。尽管转录组研究很普遍,但对于不同工作流程或“管道”对组装结果的影响了解甚少。在这里,我们通过编程自动化了一个管道,用于组装和注释作为海洋微生物真核转录组测序项目一部分收集的原始转录组短读数据。评估了由此产生的转录组组装,并将其与先前使用国家基因组研究中心开发的不同管道生成的组装进行了比较。
新的转录组组装包含了以前的大多数拼接体以及新的内容。在新组装中,平均有 7.8%的注释拼接体是以前组装中未发现的新基因名称。在组装指标中观察到了分类趋势。与其他门的转录组相比,甲藻门的组装具有更多的拼接体和独特的 k-mer,而纤毛门的组装与其他门相比,开放阅读框的比例较低。
鉴于当前的生物信息学方法,对于特定的原始数据集,没有一个单一的“最佳”参考转录组。由于最佳转录组是一个移动的目标,随着新工具和方法的不断改进(或不改进),自动化和可编程的管道对于管理重新处理具有修订管道的大量样本所需的计算密集型任务以及确保对所有样本应用通用的评估工作流程非常有价值。因此,使用自动化和可编程的管道,用新工具重新组装现有数据,除了为社区提供新的和有用的产品外,还可能更准确地识别样本中特定于分类群的趋势。