The Fred Wyszkowski Cancer Research Laboratory, Technion-Israel Institute of Technology, Haifa, Israel.
Department of Biology, Technion-Israel Institute of Technology, Haifa, Israel.
Cell Death Dis. 2018 Dec 13;9(12):1191. doi: 10.1038/s41419-018-1227-0.
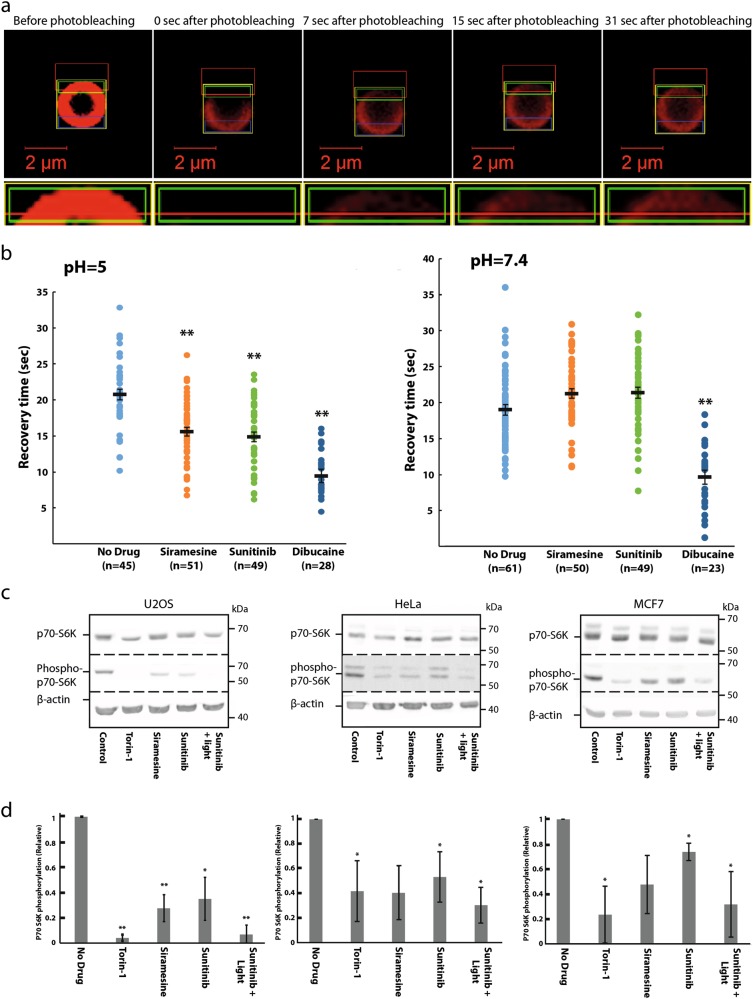
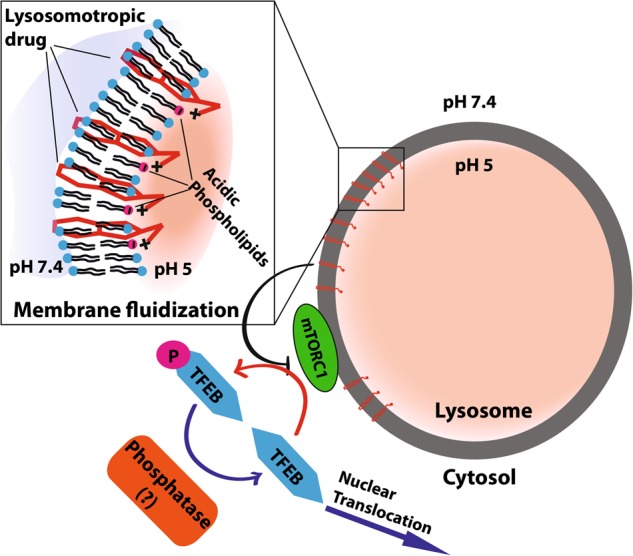
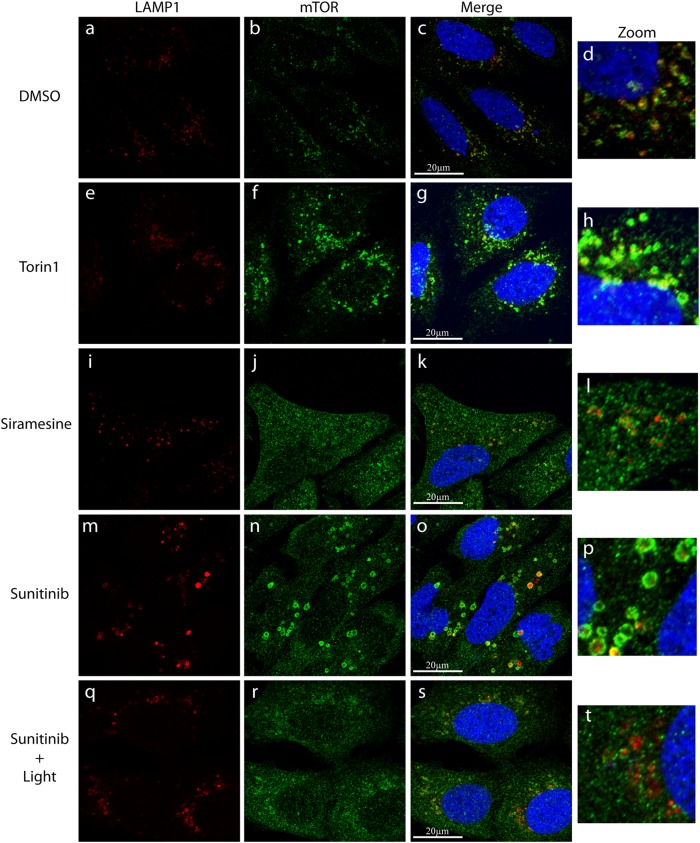
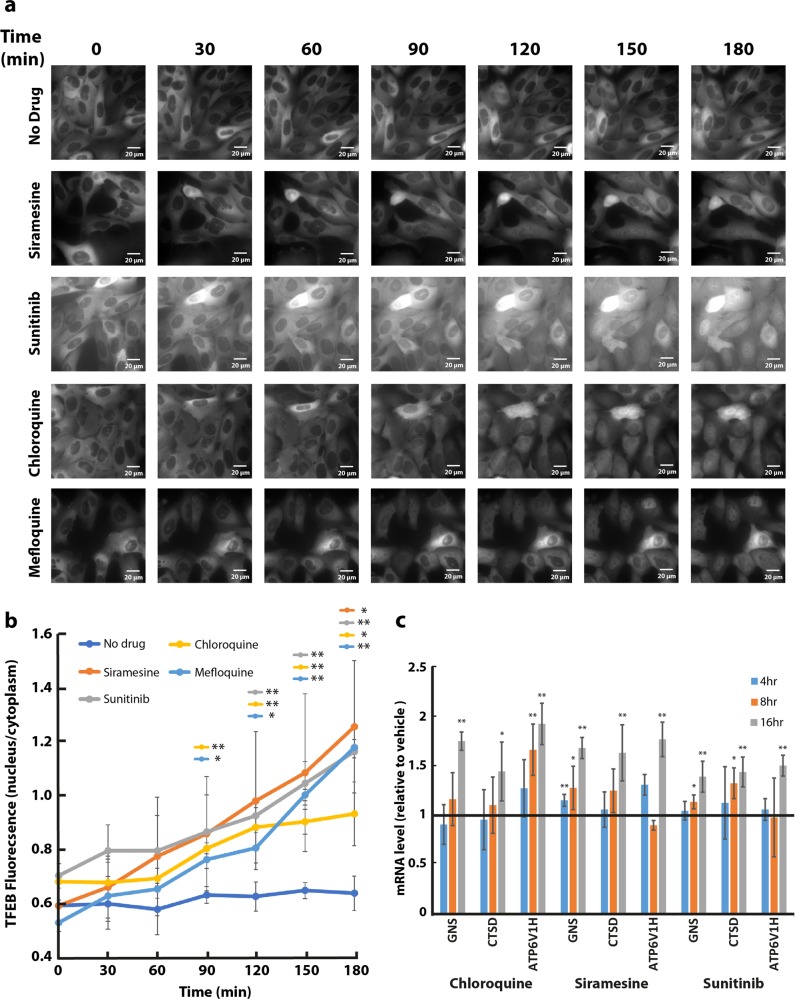
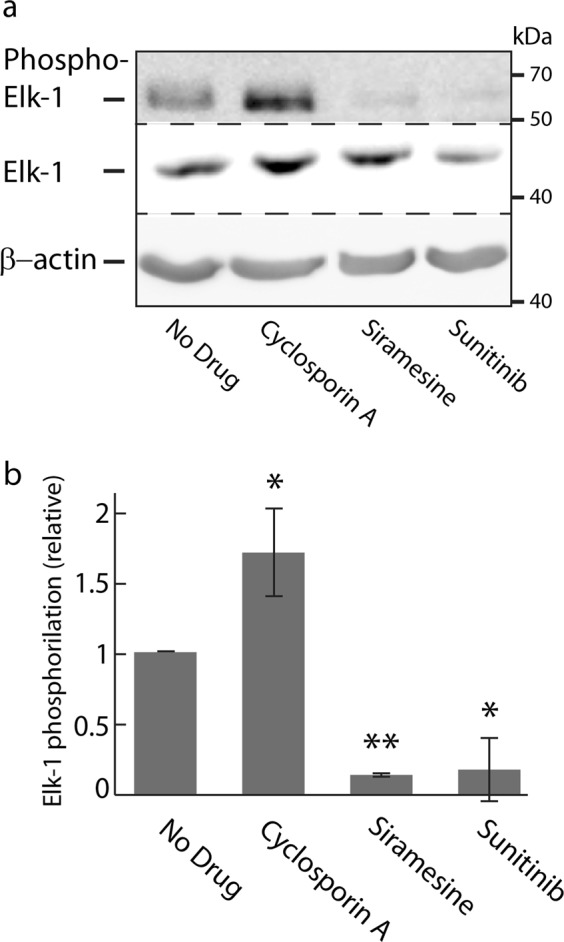
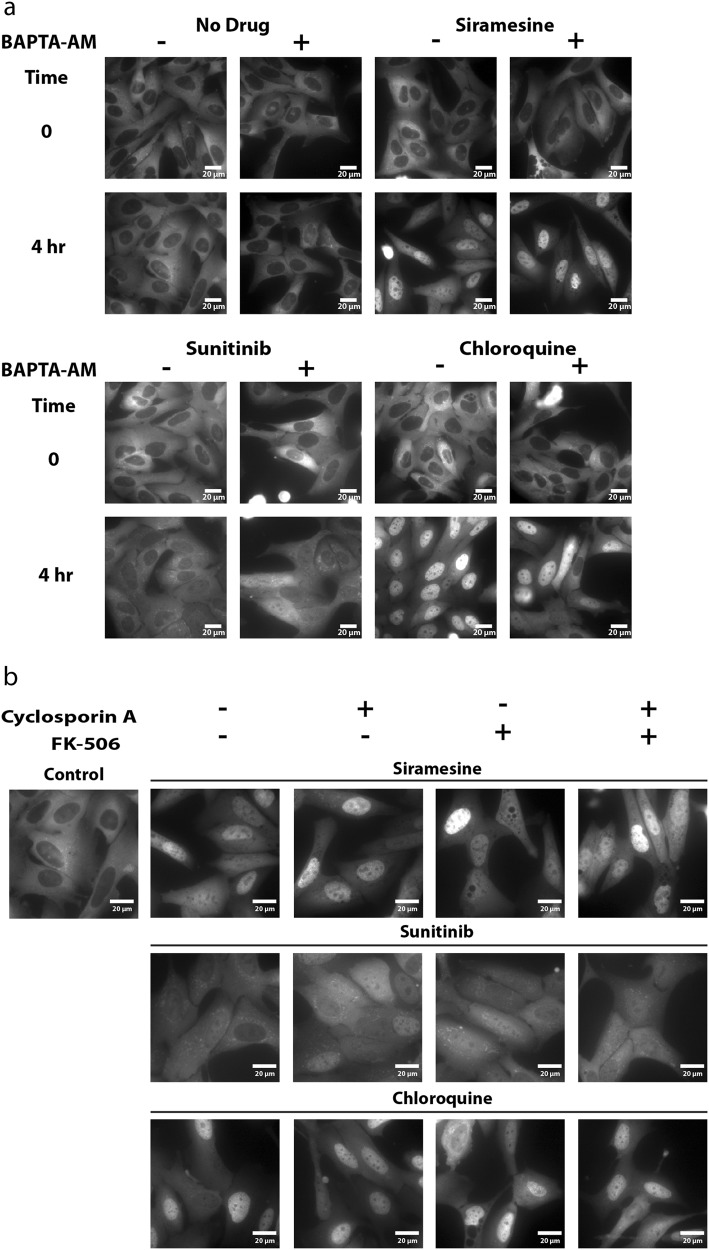
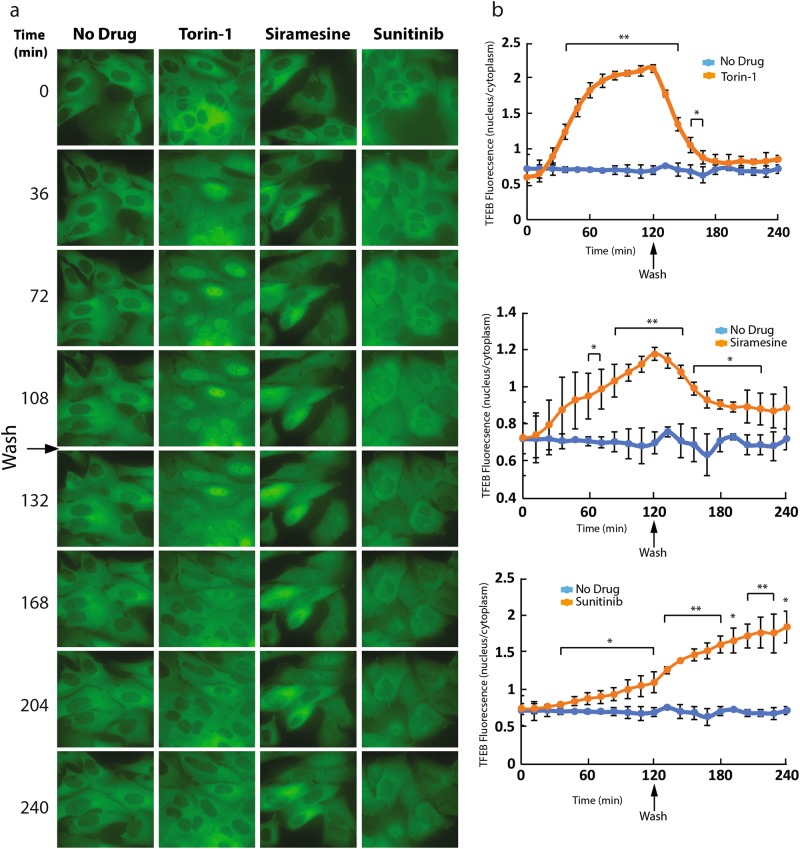
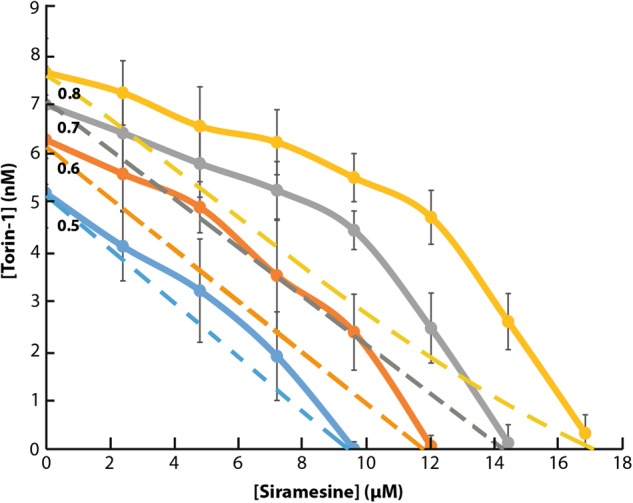
Transcription factor EB (TFEB) is a master transcriptional regulator playing a key role in lysosomal biogenesis, autophagy and lysosomal exocytosis. TFEB activity is inhibited following its phosphorylation by mammalian target of rapamycin complex 1 (mTORC1) on the surface of the lysosome. Phosphorylated TFEB is bound by 14-3-3 proteins, resulting in its cytoplasmic retention in an inactive state. It was suggested that the calcium-dependent phosphatase calcineurin is responsible for dephosphorylation and subsequent activation of TFEB under conditions of lysosomal stress. We have recently demonstrated that TFEB is activated following exposure of cancer cells to lysosomotropic anticancer drugs, resulting in lysosome-mediated cancer drug resistance via increased lysosomal biogenesis, lysosomal drug sequestration, and drug extrusion through lysosomal exocytosis. Herein, we studied the molecular mechanism underlying lysosomotropic-drug-induced activation of TFEB. We demonstrate that accumulation of lysosomotropic drugs results in membrane fluidization of lysosome-like liposomes, which is strictly dependent on the acidity of the liposomal lumen. Lysosomal accumulation of lysosomotropic drugs and the consequent fluidization of the lysosomal membrane, facilitated the dissociation of mTOR from the lysosomal membrane and inhibited the kinase activity of mTORC1, which is necessary and sufficient for the rapid translocation of TFEB to the nucleus. We further show that while lysosomotropic drug sequestration induces Ca release into the cytoplasm, facilitating calcineurin activation, chelation of cytosolic Ca, or direct inhibition of calcineurin activity, do not interfere with drug-induced nuclear translocation of TFEB. We thus suggest that lysosomotropic drug-induced activation of TFEB is mediated by mTORC1 inhibition due to lysosomal membrane fluidization and not by calcineurin activation. We further postulate that apart from calcineurin, other constitutively active phosphatase(s) partake in TFEB dephosphorylation and consequent activation. Moreover, a rapid export of TFEB from the nucleus to the cytosol occurs upon relief of mTORC1 inhibition, suggesting that dephosphorylated TFEB constantly travels between the nucleus and the cytosol, acting as a rapidly responding sensor of mTORC1 activity.
转录因子 EB(TFEB)是一种重要的转录调节因子,在溶酶体生物发生、自噬和溶酶体胞吐中发挥关键作用。TFEB 的活性在其被哺乳动物雷帕霉素靶蛋白复合物 1(mTORC1)磷酸化后受到抑制,mTORC1 位于溶酶体的表面。磷酸化的 TFEB 与 14-3-3 蛋白结合,导致其在细胞质中以非活性状态保留。有人认为,钙依赖性磷酸酶钙调神经磷酸酶负责在溶酶体应激条件下使 TFEB 去磷酸化并随后激活。我们最近证明,当癌细胞暴露于溶酶体靶向抗癌药物时,TFEB 会被激活,导致通过增加溶酶体生物发生、溶酶体药物隔离以及通过溶酶体胞吐将药物排出溶酶体,从而产生溶酶体介导的癌症药物耐药性。在此,我们研究了溶酶体靶向药物诱导 TFEB 激活的分子机制。我们证明,溶酶体靶向药物的积累导致溶酶体样脂质体的膜流化,这严格依赖于脂质体腔的酸度。溶酶体靶向药物的溶酶体积累和溶酶体膜的随后流化,促进了 mTOR 从溶酶体膜上的解离,并抑制了 mTORC1 的激酶活性,这对于 TFEB 向核的快速易位是必要和充分的。我们进一步表明,虽然溶酶体靶向药物隔离会导致细胞质中 Ca 的释放,从而促进钙调神经磷酸酶的激活,但细胞溶质 Ca 的螯合或钙调神经磷酸酶活性的直接抑制,不会干扰药物诱导的 TFEB 核易位。因此,我们认为,溶酶体靶向药物诱导的 TFEB 激活是由 mTORC1 抑制介导的,原因是溶酶体膜流化,而不是由钙调神经磷酸酶激活介导的。我们进一步假设,除了钙调神经磷酸酶之外,其他组成性激活的磷酸酶也参与了 TFEB 的去磷酸化和随后的激活。此外,当 mTORC1 抑制得到缓解时,TFEB 从核快速输出到细胞质,这表明去磷酸化的 TFEB 不断在核和细胞质之间穿梭,充当 mTORC1 活性的快速响应传感器。