1Department of Cell and Systems Biology, University of Toronto, Toronto, ON Canada.
2Department of Biochemistry, University of Toronto, Toronto, ON Canada.
NPJ Biofilms Microbiomes. 2019 Jan 21;5(1):4. doi: 10.1038/s41522-018-0077-y. eCollection 2019.
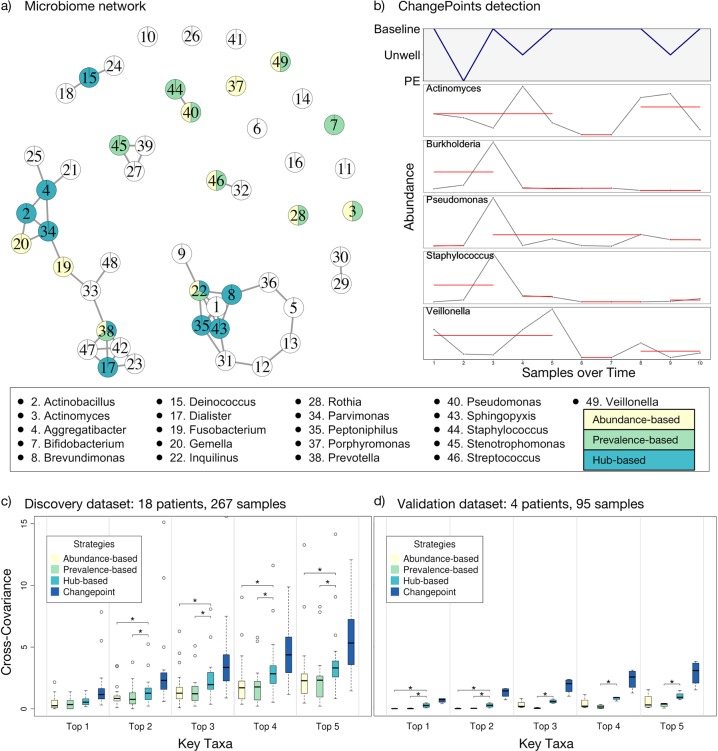
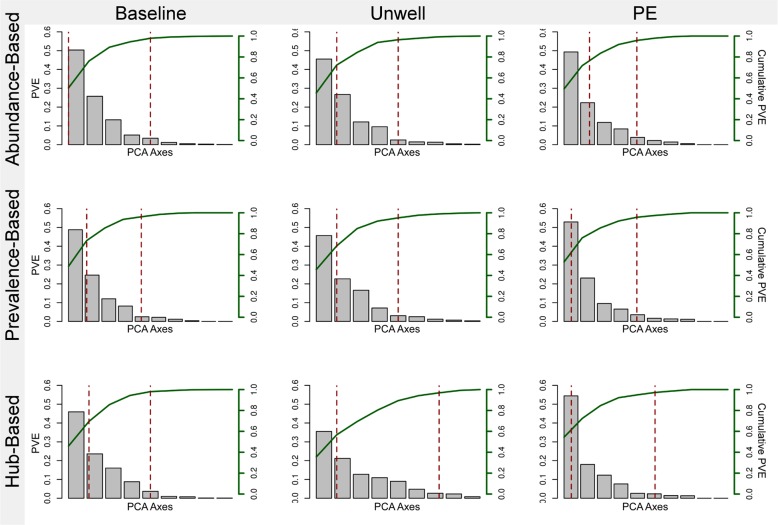
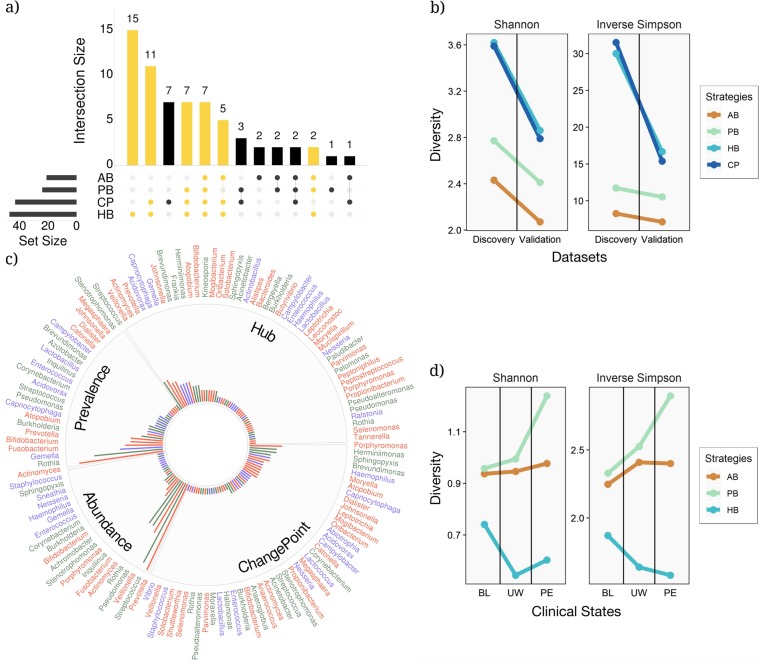
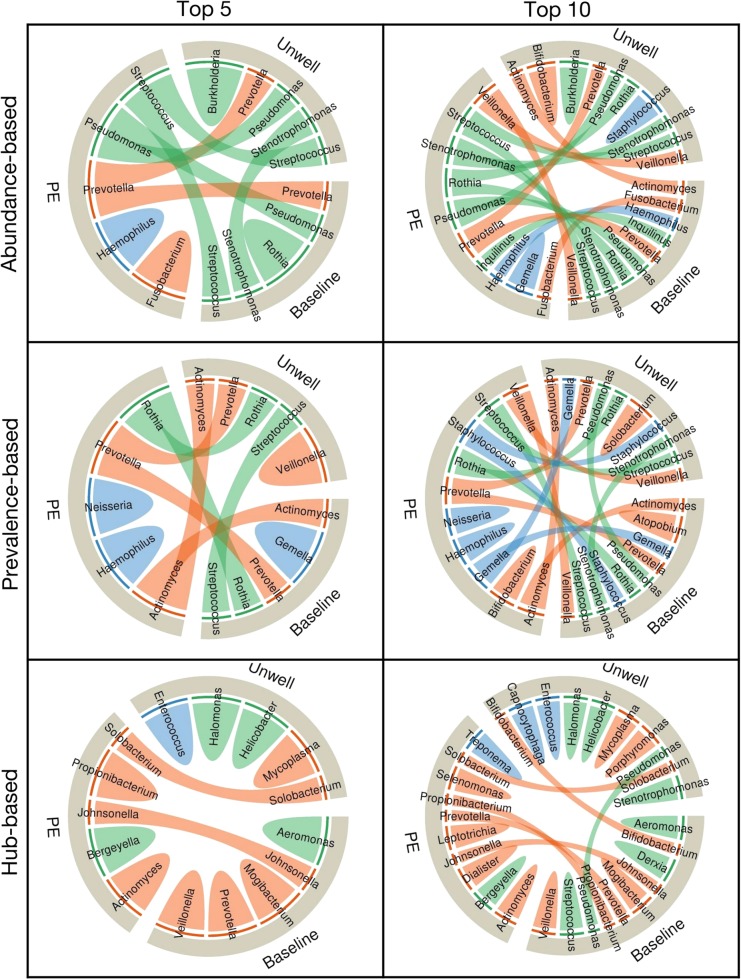
Over 90% of cystic fibrosis (CF) patients die due to chronic lung infections leading to respiratory failure. The decline in CF lung function is greatly accelerated by intermittent and progressively severe acute pulmonary exacerbations (PEs). Despite their clinical impact, surprisingly few microbiological signals associated with PEs have been identified. Here we introduce an unsupervised, systems-oriented approach to identify key members of the microbiota. We used two CF sputum microbiome data sets that were longitudinally collected through periods spanning baseline health and PEs. Key taxa were defined based on three strategies: overall relative abundance, prevalence, and co-occurrence network interconnectedness. We measured the association between changes in the abundance of the key taxa and changes in patient clinical status over time via change-point detection, and found that taxa with the highest level of network interconnectedness tracked changes in patient health significantly better than taxa with the highest abundance or prevalence. We also cross-sectionally stratified all samples into the clinical states and identified key taxa associated with each state. We found that network interconnectedness most strongly delineated the taxa among clinical states, and that anaerobic bacteria were over-represented during PEs. Many of these anaerobes are oropharyngeal bacteria that have been previously isolated from the respiratory tract, and/or have been studied for their role in CF. The observed shift in community structure, and the association of anaerobic taxa and PEs lends further support to the growing consensus that anoxic conditions and the subsequent growth of anaerobic microbes are important predictors of PEs.
超过 90%的囊性纤维化 (CF) 患者因慢性肺部感染导致呼吸衰竭而死亡。间歇性和逐渐加重的急性肺部恶化 (PE) 极大地加速了 CF 肺功能的下降。尽管它们具有临床影响,但令人惊讶的是,与 PEs 相关的微生物信号很少被识别。在这里,我们介绍了一种无监督的、面向系统的方法来识别微生物群的关键成员。我们使用了两个 CF 痰微生物组数据集,这些数据集是通过跨越基线健康和 PEs 的时期进行纵向收集的。关键分类群是基于三种策略定义的:总体相对丰度、流行率和共生网络连通性。我们通过变化点检测测量了关键分类群的丰度变化与患者临床状况随时间变化之间的关联,并发现网络连通性最高的分类群与患者健康变化的相关性明显优于丰度或流行率最高的分类群。我们还将所有样本进行横断面分层为临床状态,并确定与每种状态相关的关键分类群。我们发现,网络连通性在临床状态之间最能区分分类群,而厌氧细菌在 PEs 期间过度表达。这些厌氧菌中的许多是口腔细菌,它们以前从呼吸道中分离出来,并且/或者已经研究了它们在 CF 中的作用。观察到的群落结构变化以及厌氧分类群与 PEs 的关联进一步支持了这样一种共识,即缺氧条件和随后的厌氧微生物生长是 PEs 的重要预测因素。