Department of Molecular Life Sciences, University of Zurich, Zurich, Switzerland.
Swiss Institute of Bioinformatics, Zurich, Switzerland.
mBio. 2021 Mar 9;12(2):e02863-20. doi: 10.1128/mBio.02863-20.
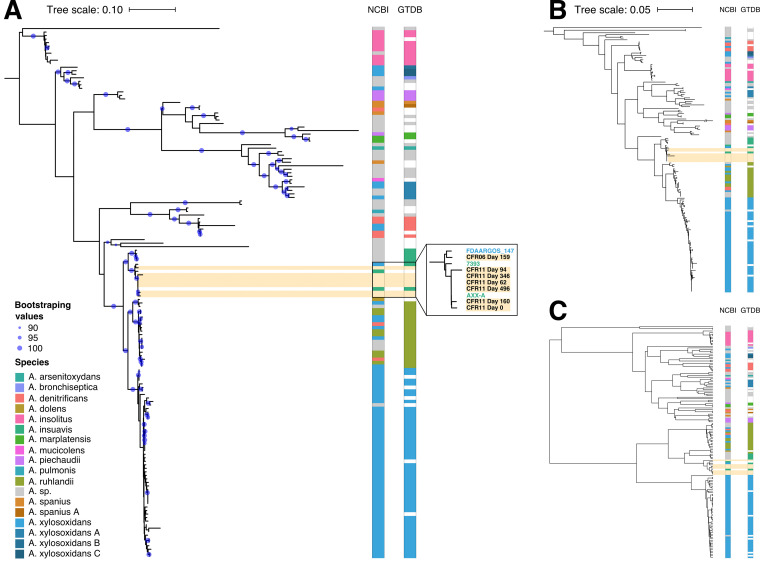
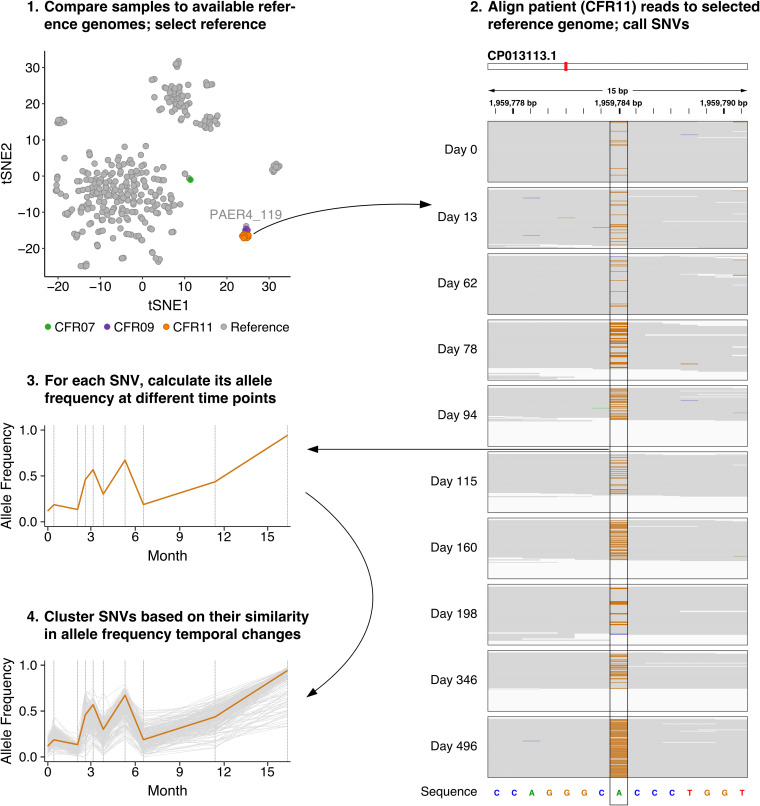
In cystic fibrosis, dynamic and complex communities of microbial pathogens and commensals can colonize the lung. Cultured isolates from lung sputum reveal high inter- and intraindividual variability in pathogen strains, sequence variants, and phenotypes; disease progression likely depends on the precise combination of infecting lineages. Routine clinical protocols, however, provide a limited overview of the colonizer populations. Therefore, a more comprehensive and precise identification and characterization of infecting lineages could assist in making corresponding decisions on treatment. Here, we describe longitudinal tracking for four cystic fibrosis patients who exhibited extreme clinical phenotypes and, thus, were selected from a pilot cohort of 11 patients with repeated sampling for more than a year. Following metagenomics sequencing of lung sputum, we find that the taxonomic identity of individual colonizer lineages can be easily established. Crucially, even superficially clonal pathogens can be subdivided into multiple sublineages at the sequence level. By tracking individual allelic differences over time, an assembly-free clustering approach allows us to reconstruct multiple lineage-specific genomes with clear structural differences. Our study showcases a culture-independent shotgun metagenomics approach for longitudinal tracking of sublineage pathogen dynamics, opening up the possibility of using such methods to assist in monitoring disease progression through providing high-resolution routine characterization of the cystic fibrosis lung microbiome. Cystic fibrosis patients frequently suffer from recurring respiratory infections caused by colonizing pathogenic and commensal bacteria. Although modern therapies can sometimes alleviate respiratory symptoms by ameliorating residual function of the protein responsible for the disorder, management of chronic respiratory infections remains an issue. Here, we propose a minimally invasive and culture-independent method to monitor microbial lung content in patients with cystic fibrosis at minimal additional effort on the patient's part. Through repeated sampling and metagenomics sequencing of our selected cystic fibrosis patients, we successfully classify infecting bacterial lineages and deconvolute multiple lineage variants of the same species within a given patient. This study explores the application of modern computational methods for deconvoluting lineages in the cystic fibrosis lung microbiome, an environment known to be inhabited by a heterogeneous pathogen population that complicates management of the disorder.
在囊性纤维化中,动态和复杂的微生物病原体和共生体群落可以定植在肺部。从肺部痰液中培养的分离株揭示了病原体菌株、序列变体和表型在个体间和个体内的高度变异性;疾病的进展可能取决于感染谱系的精确组合。然而,常规的临床方案仅提供了定植种群的有限概述。因此,更全面和精确的感染谱系鉴定和特征描述可以帮助我们在治疗方面做出相应的决策。在这里,我们描述了对四名囊性纤维化患者的纵向跟踪研究,他们表现出极端的临床表型,因此从 11 名患者的试点队列中选择,这些患者在一年以上的时间内进行了多次重复采样。在对肺部痰液进行宏基因组测序后,我们发现可以轻松确定个体定植谱系的分类学身份。至关重要的是,即使是表面上的克隆病原体,在序列水平上也可以细分为多个亚谱系。通过随时间跟踪个体等位基因差异,一种无组装的聚类方法允许我们重建具有明显结构差异的多个谱系特异性基因组。我们的研究展示了一种独立于培养的 shotgun 宏基因组学方法,用于亚谱系病原体动态的纵向跟踪,为通过提供对囊性纤维化肺部微生物组的高分辨率常规特征描述来协助监测疾病进展开辟了可能性。囊性纤维化患者经常因定植的致病性和共生细菌而遭受反复呼吸道感染。尽管现代疗法有时可以通过改善导致该疾病的蛋白质的残留功能来缓解呼吸道症状,但慢性呼吸道感染的管理仍然是一个问题。在这里,我们提出了一种微创且独立于培养的方法,以在患者最小的额外努力下监测囊性纤维化患者的肺部微生物含量。通过对我们选择的囊性纤维化患者进行重复采样和宏基因组测序,我们成功地对感染细菌谱系进行了分类,并在给定患者内解析了同一物种的多个谱系变体。这项研究探索了现代计算方法在囊性纤维化肺部微生物组中解析谱系的应用,该环境中已知存在异质病原体种群,这使得该疾病的管理变得复杂。