Lei Yunping, Kim Sung-Eun, Chen Zhongzhong, Cao Xuanye, Zhu Huiping, Yang Wei, Shaw Gary M, Zheng Yufang, Zhang Ting, Wang Hong-Yan, Finnell Richard H
Department of Nutritional Sciences, Dell Pediatric Research Institute, University of Texas at Austin Dell Medical School, Austin, Texas.
Obstetrics and Gynecology Hospital, State Key Laboratory of Genetic Engineering at School of Life Sciences, Institute of Reproduction and Development, Fudan University, Shanghai, China.
Mol Genet Genomic Med. 2019 Apr;7(4):e00584. doi: 10.1002/mgg3.584. Epub 2019 Jan 28.
Variants in planar cell polarity (PCP) pathway genes have been repeatedly implicated in the pathogenesis of NTDs in both mouse models and in human cohorts. Mouse models indicate that the homogenous disruption of the Ptk7 gene, a PCP regulator, results in craniorachischisis; while embryos that are doubly heterozygous for Ptk7 and Vangl2 mutations present with spina bifida.
In this study, we initially sequenced exons of the human PTK7 gene in 192 spina bifida patients and 190 controls from a California population. A phase II validation study was performed in 343 Chinese NTD cohort. Functional assays including immunoblotting and immunoprecipitation were used to study identified variants effect on PTK7 function.
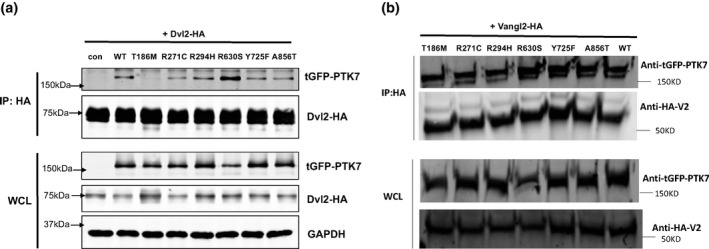
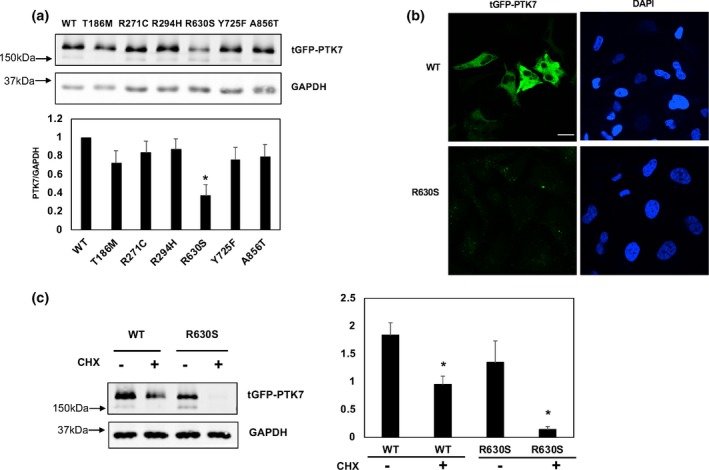
We identified three rare (MAF <0.001) missense heterozygous PTK7 variants (NM_001270398.1:c.581C>T, p.Arg630Ser and p.Tyr725Phe) in the spina bifida patients. In our functional analyses, p.Arg630Ser affected PTK7 mutant protein stability and increased interaction with Dvl2, while the p.Thr186Met variant decreased PTK7 interactions with Dvl2. No novel predicted-to-be-damaging variant or function-disrupted PTK7 variant was identified among the control subjects. We subsequently re-sequenced the PTK7 CDS region in 343 NTDs from China to validate the association between PTK7 and NTDs. The frequency of PTK7 rare missense variants in the Chinese NTD samples is significantly higher than in gnomAD controls.
Our study suggests that rare missense variants in PTK7 contribute to the genetic risk of NTDs.
平面细胞极性(PCP)通路基因的变异在小鼠模型和人类队列研究中均反复被认为与神经管缺陷(NTDs)的发病机制有关。小鼠模型表明,PCP调节因子Ptk7基因的同质破坏会导致脊柱裂;而Ptk7和Vangl2基因双杂合突变的胚胎则表现为脊柱裂。
在本研究中,我们首先对来自加利福尼亚州人群的192例脊柱裂患者和190例对照者的人类PTK7基因外显子进行了测序。在343例中国NTD队列中进行了二期验证研究。采用免疫印迹和免疫沉淀等功能分析方法,研究已鉴定变异对PTK7功能的影响。
我们在脊柱裂患者中鉴定出三个罕见的(MAF<0.001)错义杂合PTK7变异(NM_001270398.1:c.581C>T,p.Arg630Ser和p.Tyr725Phe)。在我们的功能分析中,p.Arg630Ser影响PTK7突变蛋白的稳定性,并增加与Dvl2的相互作用,而p.Thr186Met变异则降低PTK7与Dvl2的相互作用。在对照受试者中未鉴定出新的预测有害变异或功能破坏的PTK7变异。随后,我们对来自中国的343例NTD患者的PTK7编码区进行了重新测序,以验证PTK7与NTDs之间的关联。中国NTD样本中PTK7罕见错义变异的频率显著高于gnomAD对照。
我们的研究表明,PTK7中的罕见错义变异会增加NTDs的遗传风险。