Department of Population and Quantitative Health Science, School of Medicine, Case Western Reserve University, Cleveland, OH, 44106, USA.
BMC Genomics. 2019 Feb 11;20(1):124. doi: 10.1186/s12864-019-5510-y.
Rheumatoid arthritis (RA) is the most common autoimmune disease and affects about 1% of the population. The cause of RA remains largely unknown and could result from a complex interaction between genes and environment factors. Recent studies suggested that gut microbiota and their collective metabolic outputs exert profound effects on the host immune system and are implicated in RA. However, which and how gut microbial metabolites interact with host genetics in contributing to RA pathogenesis remains unknown. In this study, we present a data-driven study to understand how gut microbial metabolites contribute to RA at the genetic, functional and phenotypic levels.
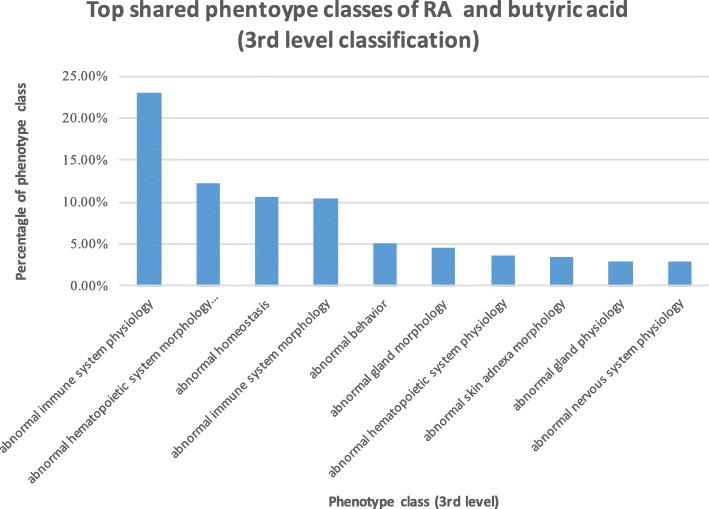
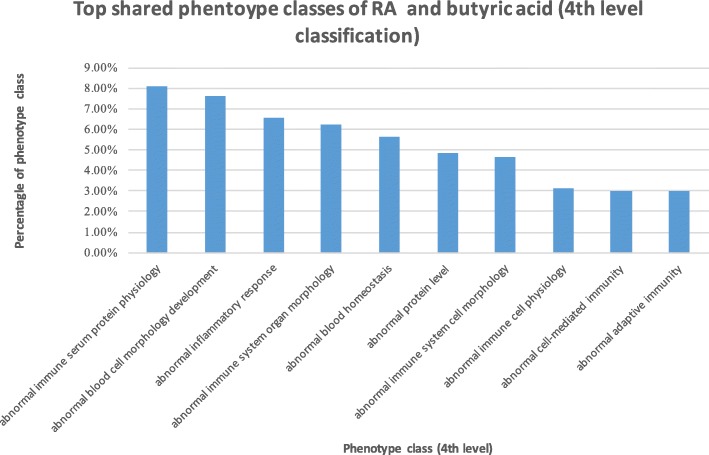
We used publicly available disease genetics, chemical genetics, human metabolome, genetic signaling pathways, mouse genome-wide mutation phenotypes, and mouse phenotype ontology data. We identified RA-associated microbial metabolites and prioritized them based on their genetic, functional and phenotypic relevance to RA. We evaluated the prioritization methods using short-chain fatty acids (SCFAs), which were previously shown to be involved in RA etiology. We validate the algorithms by showing that SCFAs are highly associated with RA at genetic, functional and phenotypic levels: SCFAs ranked at top 3.52% based on shared genes with RA, top 5.69% based on shared genetic pathways, and top 16.94% based on shared phenotypes. Based on the genetic-level analysis, human gut microbial metabolites directly interact with many RA-associated genes (as many as 18.1% of all 166 RA genes). Based on the functional-level analysis, human gut microbial metabolites participate in many RA-associated genetic pathways (as many as 71.4% of 311 genetic pathways significantly enriched for RA), including immune system pathways. Based on the phenotypic-level analysis, gut microbial metabolites affect many RA-related phenotypes (as many as 51.3% of 978 phenotypes significantly enriched for RA), including many immune system phenotypes.
Our study demonstrates strong gut-microbiome-immune-joint interactions in RA, which converged on both genetic, functional and phenotypic levels.
类风湿关节炎(RA)是最常见的自身免疫性疾病,影响约 1%的人口。RA 的病因在很大程度上仍然未知,可能是由基因和环境因素的复杂相互作用引起的。最近的研究表明,肠道微生物群及其集体代谢产物对宿主免疫系统有深远的影响,并与 RA 有关。然而,肠道微生物代谢物如何与宿主遗传相互作用,从而导致 RA 的发病机制仍不清楚。在这项研究中,我们提出了一项数据驱动的研究,以了解肠道微生物代谢物如何在遗传、功能和表型水平上导致 RA。
我们使用了公开的疾病遗传学、化学遗传学、人类代谢组学、遗传信号通路、小鼠全基因组突变表型和小鼠表型本体数据。我们确定了与 RA 相关的微生物代谢物,并根据它们与 RA 的遗传、功能和表型相关性对其进行了优先级排序。我们使用先前被证明与 RA 病因学有关的短链脂肪酸(SCFA)来评估优先级排序方法。我们通过证明 SCFA 在遗传、功能和表型水平上与 RA 高度相关来验证算法:根据与 RA 共享的基因,SCFA 排在前 3.52%;根据共享的遗传途径,排在前 5.69%;根据共享的表型,排在前 16.94%。基于遗传水平的分析,人类肠道微生物代谢物直接与许多 RA 相关的基因相互作用(多达 18.1%的 166 个 RA 基因)。基于功能水平的分析,人类肠道微生物代谢物参与了许多与 RA 相关的遗传途径(多达 311 个遗传途径中有 71.4%与 RA 显著富集),包括免疫系统途径。基于表型水平的分析,肠道微生物代谢物影响许多与 RA 相关的表型(多达 978 个表型中有 51.3%与 RA 显著富集),包括许多免疫系统表型。
我们的研究表明,RA 中存在强烈的肠道微生物-免疫-关节相互作用,这些作用在遗传、功能和表型水平上都有体现。