Department of Cancer Biology and Genetics, The Ohio State University Comprehensive Cancer Center, Columbus, OH, 43210, USA.
Dipartimento di Medicina Sperimentale e Clinica, University "Magna Græcia" of Catanzaro, Catanzaro, 88100, Italy.
Cell Death Dis. 2019 Feb 15;10(3):147. doi: 10.1038/s41419-019-1414-7.
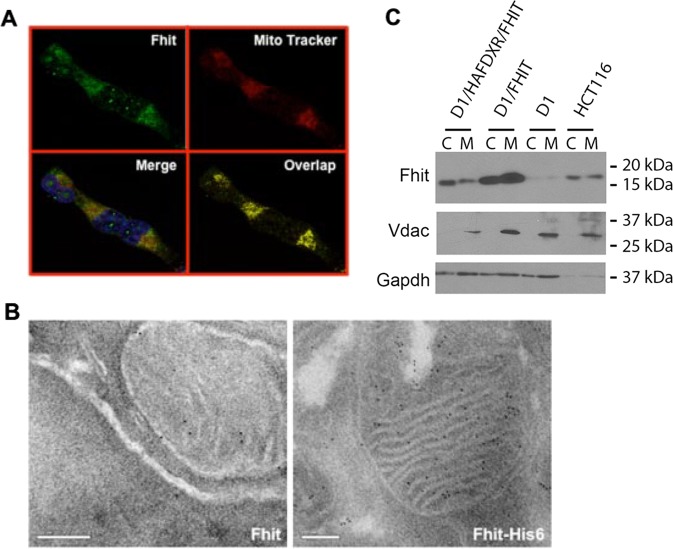
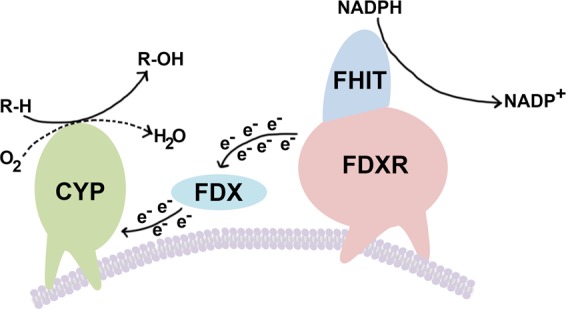
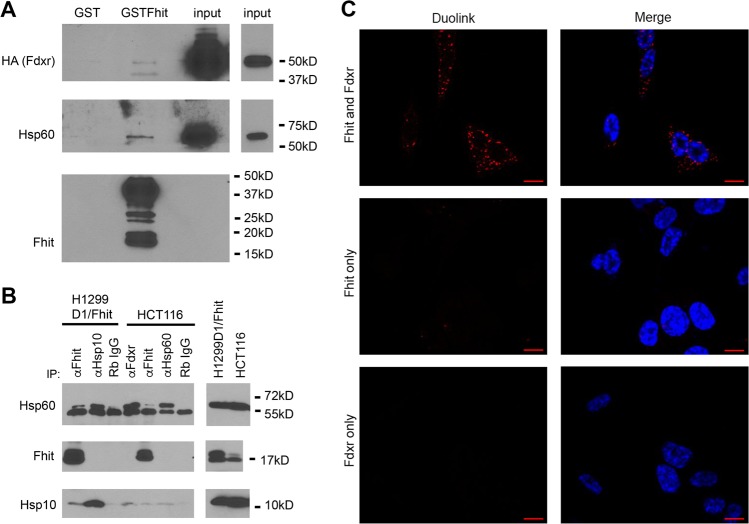
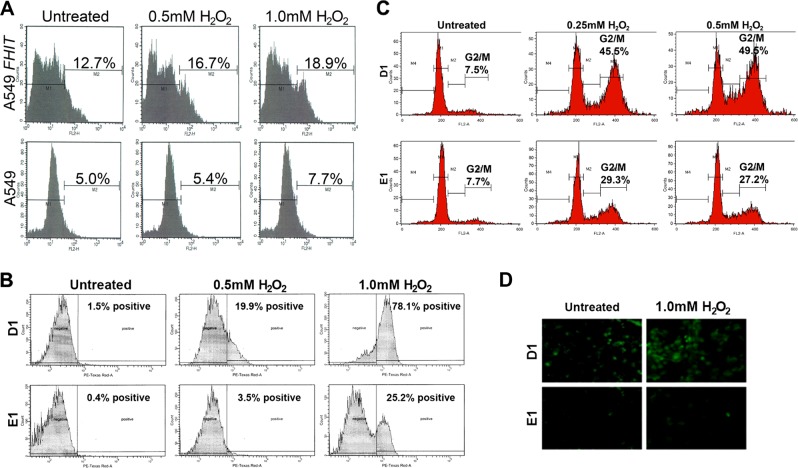
Fhit protein is lost in cancers of most, perhaps all, cancer types; when restored, it can induce apoptosis and suppress tumorigenicity, as shown in vitro and in mouse tumor models in vivo. Following protein cross-linking and proteomics analyses, we characterized a Fhit protein complex involved in triggering Fhit-mediated apoptosis. The complex includes the heat-shock chaperonin pair, HSP60/10, which is likely involved in importing Fhit into the mitochondria, where it interacts with ferredoxin reductase, responsible for transferring electrons from NADPH to cytochrome P450 via ferredoxin, in electron transport chain complex III. Overexpression of Fhit protein in Fhit-deficient cancer cells modulates the production of intracellular reactive oxygen species, causing increased ROS, following peroxide treatment, with subsequent increased apoptosis of lung cancer cells under oxidative stress conditions; conversely, Fhit-negative cells escape ROS overproduction and ROS-induced apoptosis, likely carrying oxidative damage. Thus, characterization of Fhit-interacting proteins has identified direct effectors of a Fhit-mediated apoptotic signal pathway that is lost in many cancers. This is of translational interest considering the very recent emphasis in a number of high-profile publications, concerning the role of oxidative phosphorylation in the treatment of human cancers, and especially cancer stem cells that rely upon oxidative phosphorylation for survival. Additionally, we have shown that cells from a Fhit-deficient lung cancer cell line, are sensitive to killing by exposure to atovaquone, thought to act as a selective oxidative phosphorylation inhibitor by targeting the CoQ10 dependence of the mitochondrial complex III, while the Fhit-expressing sister clone is resistant to this treatment.
脆性组氨酸三联体(Fhit)蛋白在大多数(如果不是全部)癌症类型的癌症中丢失;当恢复时,它可以诱导细胞凋亡并抑制肿瘤发生,这已在体外和体内小鼠肿瘤模型中得到证实。通过蛋白质交联和蛋白质组学分析,我们鉴定了一个参与触发 Fhit 介导的细胞凋亡的 Fhit 蛋白复合物。该复合物包括热休克伴侣蛋白对 HSP60/10,它可能参与将 Fhit 导入线粒体,在那里它与铁氧还蛋白还原酶相互作用,后者负责通过铁氧还蛋白将电子从 NADPH 转移到细胞色素 P450,在电子传递链复合物 III 中。在 Fhit 缺陷型癌细胞中过表达 Fhit 蛋白可调节细胞内活性氧的产生,导致过氧化物处理后 ROS 增加,随后在氧化应激条件下肺癌细胞的凋亡增加;相反,Fhit 阴性细胞逃避 ROS 过度产生和 ROS 诱导的细胞凋亡,可能携带氧化损伤。因此,鉴定 Fhit 相互作用蛋白的特征确定了 Fhit 介导的细胞凋亡信号通路的直接效应物,该信号通路在许多癌症中丢失。考虑到最近在一些高知名度的出版物中强调氧化磷酸化在人类癌症治疗中的作用,特别是依赖氧化磷酸化生存的癌症干细胞,这具有转化意义。此外,我们已经表明,来自 Fhit 缺陷型肺癌细胞系的细胞对阿托伐醌的杀伤敏感,阿托伐醌被认为通过靶向线粒体复合物 III 的 CoQ10 依赖性来作为一种选择性氧化磷酸化抑制剂,而表达 Fhit 的姐妹克隆对这种治疗具有抗性。