Science for Life Laboratory, Division of Gene Technology, School of Engineering Sciences in Chemistry, Biotechnology and Health, KTH Royal Institute of Technology, Solna, Sweden (Ö.Å., R.S., S.P., A.A., P.H., P.S.).
Cardiovascular Medicine Unit, Department of Medicine, Center for Molecular Medicine, Karolinska Institutet, Stockholm, Sweden (F.-A.P., P.E.).
Circ Genom Precis Med. 2019 Mar;12(3):e002353. doi: 10.1161/CIRCGEN.118.002353.
Genetic variant landscape of coronary artery disease is dominated by noncoding variants among which many occur within putative enhancers regulating the expression levels of relevant genes. It is crucial to assign the genetic variants to their correct genes both to gain insights into perturbed functions and better assess the risk of disease.
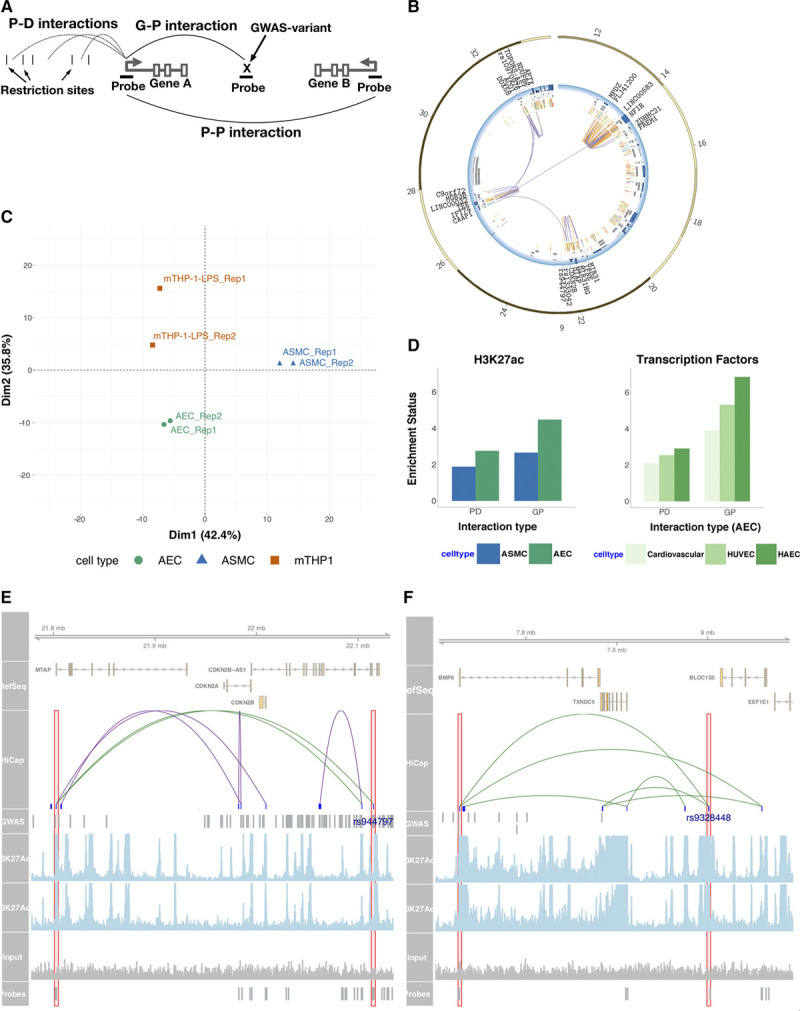
In this study, we generated high-resolution genomic interaction maps (≈750 bases) in aortic endothelial, smooth muscle cells and THP-1 (human leukemia monocytic cell line) macrophages stimulated with lipopolysaccharide using Hi-C coupled with sequence capture targeting 25 429 features, including variants associated with coronary artery disease. We also sequenced their transcriptomes and mapped putative enhancers using chromatin immunoprecipitation with an antibody against H3K27Ac.
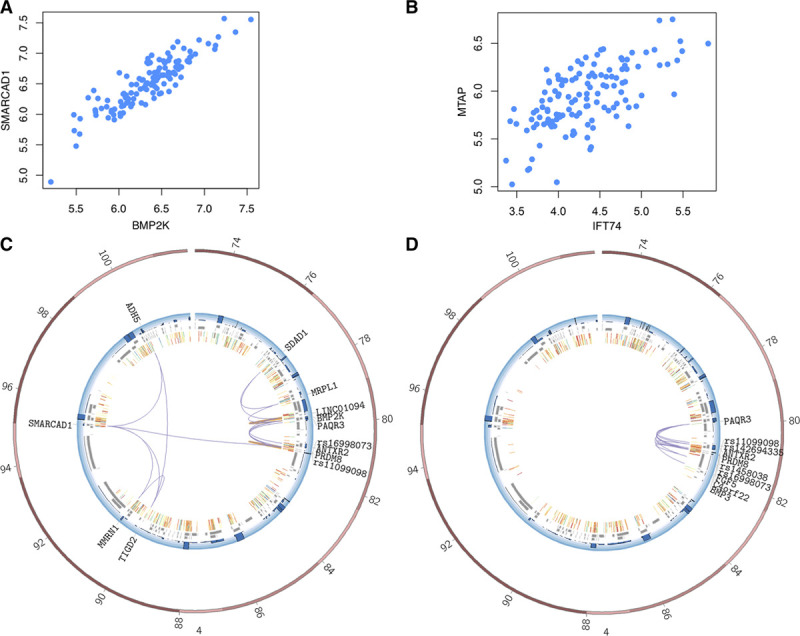
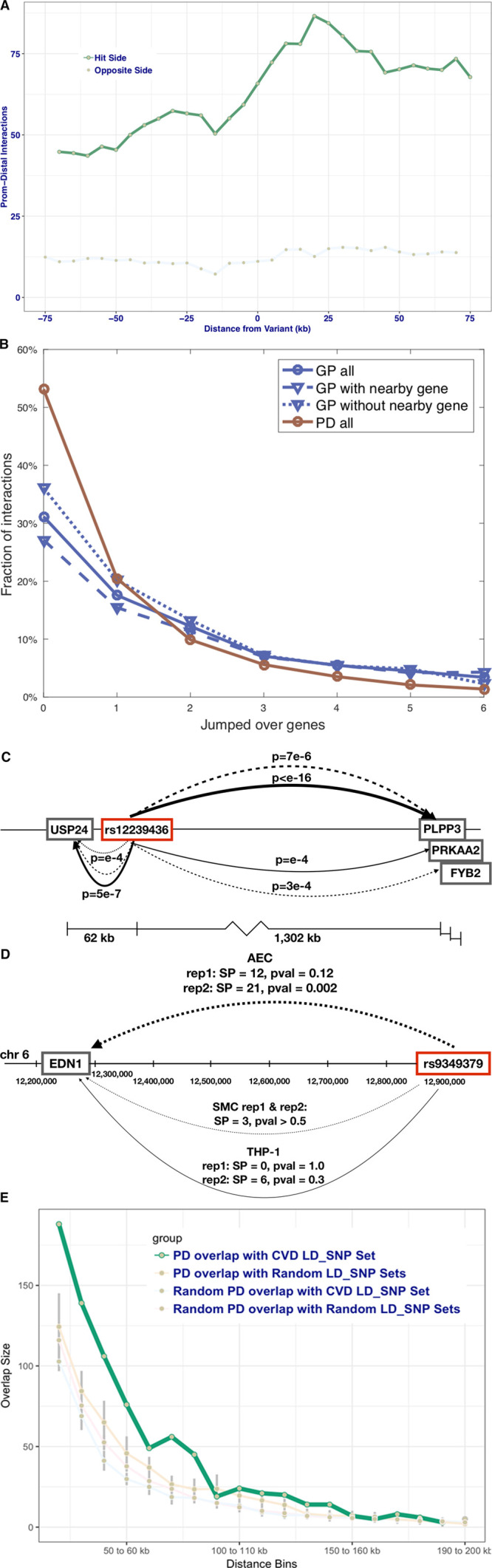
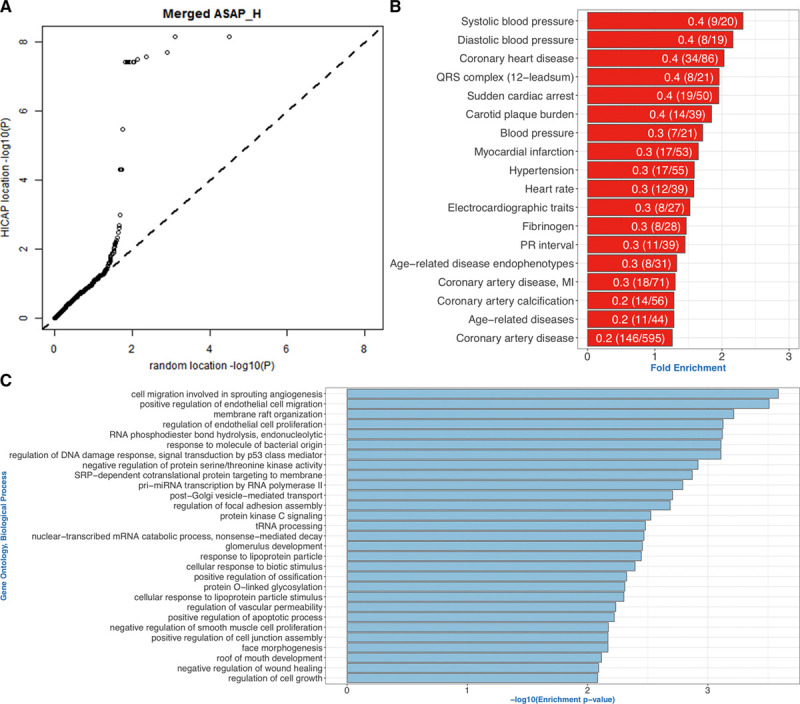
The regions interacting with promoters showed strong enrichment for enhancer elements and validated several previously known interactions and enhancers. We detected interactions for 727 risk variants obtained by genome-wide association studies and identified novel, as well as established genes and functions associated with cardiovascular diseases. We were able to assign potential target genes for additional 398 genome-wide association studies variants using haplotype information, thereby identifying additional relevant genes and functions. Importantly, we discovered that a subset of risk variants interact with multiple promoters and their expression levels were strongly correlated.
In summary, we present a catalog of candidate genes regulated by coronary artery disease-related variants and think that it will be an invaluable resource to further the investigation of cardiovascular pathologies and disease.
冠心病的遗传变异景观主要由非编码变异主导,其中许多变异发生在调节相关基因表达水平的假定增强子中。将遗传变异分配到正确的基因中至关重要,这既能深入了解受干扰的功能,又能更好地评估疾病风险。
在这项研究中,我们使用 Hi-C 结合靶向 25429 个特征(包括与冠心病相关的变异)的序列捕获,在经脂多糖刺激的主动脉内皮细胞、平滑肌细胞和 THP-1(人白血病单核细胞系)巨噬细胞中生成了高分辨率的基因组相互作用图谱(≈750 个碱基对)。我们还对其转录组进行了测序,并使用针对 H3K27Ac 的染色质免疫沉淀来绘制假定的增强子。
与启动子相互作用的区域强烈富集了增强子元件,并验证了几个先前已知的相互作用和增强子。我们检测到 727 个通过全基因组关联研究获得的风险变异的相互作用,并鉴定了与心血管疾病相关的新的和已建立的基因和功能。我们能够使用单倍型信息为另外 398 个全基因组关联研究变体分配潜在的靶基因,从而确定了额外的相关基因和功能。重要的是,我们发现一部分风险变异与多个启动子相互作用,并且它们的表达水平高度相关。
总之,我们提出了一个由冠心病相关变异调控的候选基因目录,我们认为这将是进一步研究心血管病理和疾病的宝贵资源。