From the ‡The Broad Institute or MIT and Harvard, Cambridge, Massachusetts 02142;; §Klarman Cell Observatory, The Broad Institute of MIT and Harvard, Cambridge, Massachusetts 02142;.
¶Division of Immunology, Department of Microbiology and Immunobiology, Harvard Medical School, and Evergrande Center for Immunologic Diseases, Harvard Medical School and Brigham and Women's Hospital, Boston, Massachusetts.
Mol Cell Proteomics. 2019 May;18(5):995-1009. doi: 10.1074/mcp.RA118.001259. Epub 2019 Feb 21.
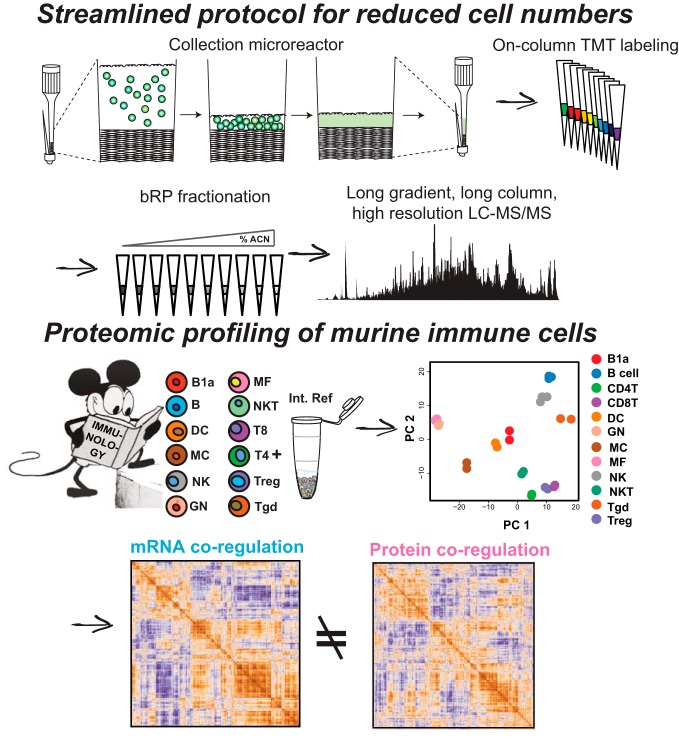
Proteomic profiling describes the molecular landscape of proteins in cells immediately available to sense, transduce, and enact the appropriate responses to extracellular queues. Transcriptional profiling has proven invaluable to our understanding of cellular responses; however, insights may be lost as mounting evidence suggests transcript levels only moderately correlate with protein levels in steady state cells. Mass spectrometry-based quantitative proteomics is a well-suited and widely used analytical tool for studying global protein abundances. Typical proteomic workflows are often limited by the amount of sample input that is required for deep and quantitative proteome profiling. This is especially true if the cells of interest need to be purified by fluorescence-activated cell sorting (FACS) and one wants to avoid culturing. To address this need, we developed an easy to implement, streamlined workflow that enables quantitative proteome profiling from roughly 2 μg of protein input per experimental condition. Utilizing a combination of facile cell collection from cell sorting, solid-state isobaric labeling and multiplexing of peptides, and small-scale fractionation, we profiled the proteomes of 12 freshly isolated, primary murine immune cell types. Analyzing half of the 3e5 cells collected per cell type, we quantified over 7000 proteins across 12 key immune cell populations directly from their resident tissues. We show that low input proteomics is precise, and the data generated accurately reflects many aspects of known immunology, while expanding the list of cell-type specific proteins across the cell types profiled. The low input proteomics methods we developed are readily adaptable and broadly applicable to any cell or sample types and should enable proteome profiling in systems previously unattainable.
蛋白质组学描述了细胞中立即可用于感知、转导和实施对外界队列的适当反应的蛋白质的分子图谱。转录组学 profiling 已被证明对我们理解细胞反应非常有价值;然而,越来越多的证据表明,转录水平与稳定状态细胞中的蛋白质水平仅中度相关,这可能会导致见解的丧失。基于质谱的定量蛋白质组学是研究全局蛋白质丰度的一种非常适合和广泛使用的分析工具。典型的蛋白质组学工作流程通常受到需要进行深度和定量蛋白质组学分析的样品输入量的限制。如果感兴趣的细胞需要通过荧光激活细胞分选 (FACS) 进行纯化,并且希望避免培养,那么情况尤其如此。为了解决这一需求,我们开发了一种易于实施的简化工作流程,该流程可从每个实验条件约 2μg 的蛋白质输入中进行定量蛋白质组学分析。我们利用从细胞分选中方便地收集细胞、固态等压标记和肽的多路复用以及小规模分级分离的组合,对 12 种新鲜分离的、主要的小鼠免疫细胞类型的蛋白质组进行了分析。分析每个细胞类型收集的一半 3e5 细胞,我们直接从它们的常驻组织中对 12 种关键免疫细胞群体中的超过 7000 种蛋白质进行了定量。我们表明,低输入蛋白质组学是精确的,生成的数据准确反映了已知免疫学的许多方面,同时扩展了所分析细胞类型的细胞类型特异性蛋白质列表。我们开发的低输入蛋白质组学方法易于适应并且广泛适用于任何细胞或样品类型,应该能够在以前无法达到的系统中进行蛋白质组学分析。