Kwon Minji, Seo Sang-Soo, Kim Mi Kyung, Lee Dong Ock, Lim Myoung Cheol
Division of Cancer Epidemiology and Prevention, National Cancer Center, 323, Ilsan-ro, Ilsandong-gu, Goyang-si 10408, Korea.
Center for Uterine Cancer, National Cancer Center, 323, Ilsan-ro, Ilsandong-gu, Goyang-si 10408, Korea.
Cancers (Basel). 2019 Mar 5;11(3):309. doi: 10.3390/cancers11030309.
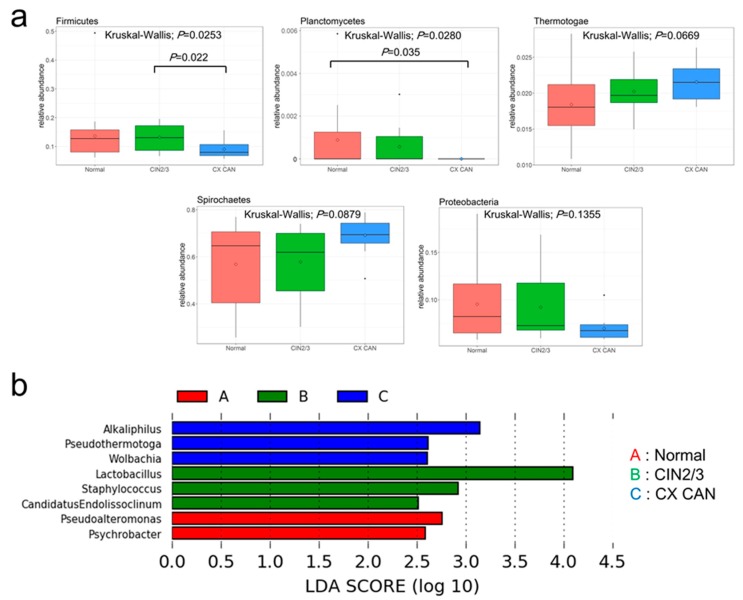
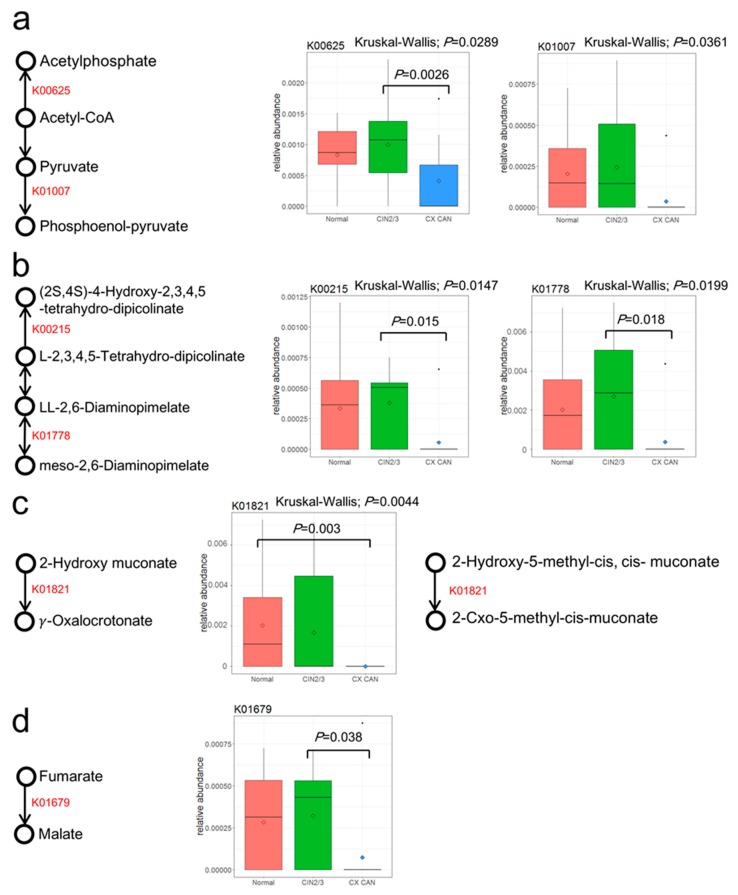
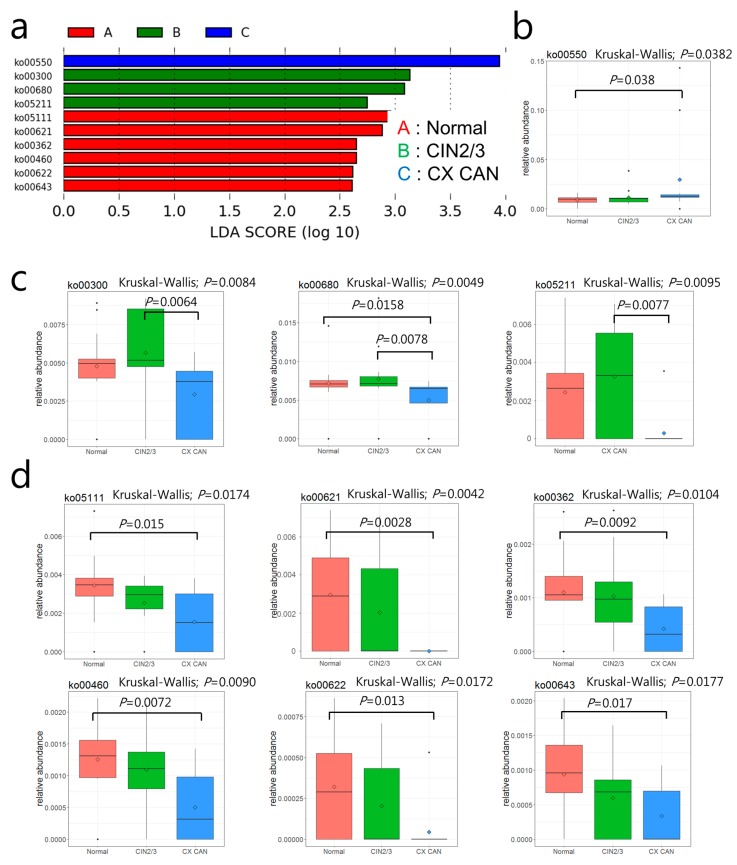
Recent studies have reported the potential role of microbiomes in cervical disease. However, little is known about the microbiome composition and function in cervical carcinogenesis. We aimed to identify the compositional and functional alterations of cervical microbiomes in cases of cervical carcinogenesis of Korean women using shotgun metagenomic sequencing. In this study, using shotgun sequencing, we sequenced the cervical metagenomes of cervical intraneoplasia 2/3 ( = 17), cervical cancer ( = 12), and normal controls ( = 18) to identify the microbial abundances and enriched metabolic functions in cervical metagenomes. At the genus level, the microbiota of cervical cancer were differentially enriched with genera , , and . Cervical intraepithelial neoplasia (CIN) 2/3 were enriched with , and . The normal group was enriched with and . Further characterization of the functionalities of the metagenomes may suggest that six Kyoto Encyclopedia of Genes and Genomes (KEGG) orthologies (KOs) that are involved in 10 pathways are associated with an increased risk of CIN2/3 and cervical cancer. Specifically, cervical metagenomes were enriched in the course of peptidoglycan synthesis and depleted by dioxin degradation and 4-oxalocrotonate tautomerase. The Cluster of Orthologous Groups (COG) category 'Defense mechanisms' was depleted in cervical cancer patients. Our findings based on shotgun metagenomic sequencing suggest that cervical microbiome community compositions and their metagenomics profiles differed between cervical lesions and normal subjects. Future studies should have larger sample sizes and/or aggregate their results to have sufficient power to detect reproducible and significant associations.
近期研究报道了微生物群在宫颈疾病中的潜在作用。然而,关于微生物群在宫颈癌发生过程中的组成和功能却知之甚少。我们旨在通过鸟枪法宏基因组测序确定韩国女性宫颈癌发生病例中宫颈微生物群的组成和功能改变。在本研究中,我们使用鸟枪法测序对宫颈上皮内瘤变2/3(n = 17)、宫颈癌(n = 12)和正常对照(n = 18)的宫颈宏基因组进行测序,以确定宫颈宏基因组中的微生物丰度和富集的代谢功能。在属水平上,宫颈癌的微生物群在属、 和 中差异富集。宫颈上皮内瘤变(CIN)2/3在 、 和 中富集。正常组在 和 中富集。对宏基因组功能的进一步表征可能表明,参与10条途径的6个京都基因和基因组百科全书(KEGG)直系同源物(KO)与CIN2/3和宫颈癌风险增加相关。具体而言,宫颈宏基因组在肽聚糖合成过程中富集,而在二恶英降解和4-氧代巴豆酸互变异构酶作用下减少。直系同源簇(COG)类别“防御机制”在宫颈癌患者中减少。我们基于鸟枪法宏基因组测序的研究结果表明,宫颈病变和正常受试者之间的宫颈微生物群群落组成及其宏基因组学特征存在差异。未来的研究应该有更大的样本量和/或汇总其结果,以有足够的能力检测可重复和显著的关联。