Peng Weijun, Huang Jianhua, Yang Jingjing, Zhang Zheyu, Yu Rong, Fayyaz Sharmeen, Zhang Shuihan, Qin Yu-Hui
Department of Integrated Traditional Chinese and Western Medicine, The Second Xiangya Hospital, Central South University, Changsha, China.
Hunan Academy of Chinese Medicine, Hunan University of Chinese Medicine, Changsha, China.
Front Microbiol. 2020 Jan 20;10:3141. doi: 10.3389/fmicb.2019.03141. eCollection 2019.
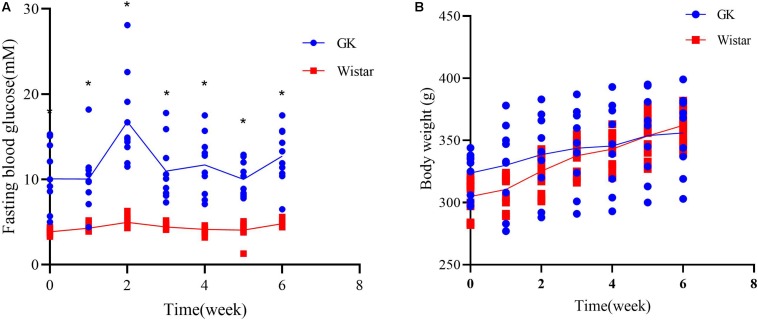
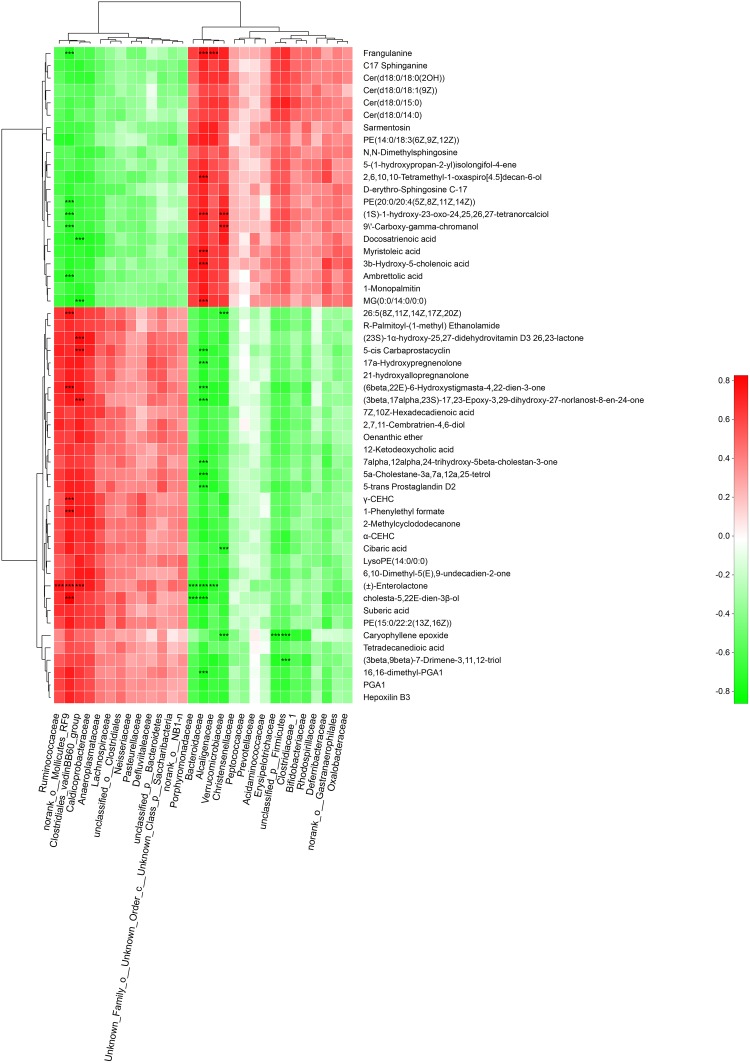
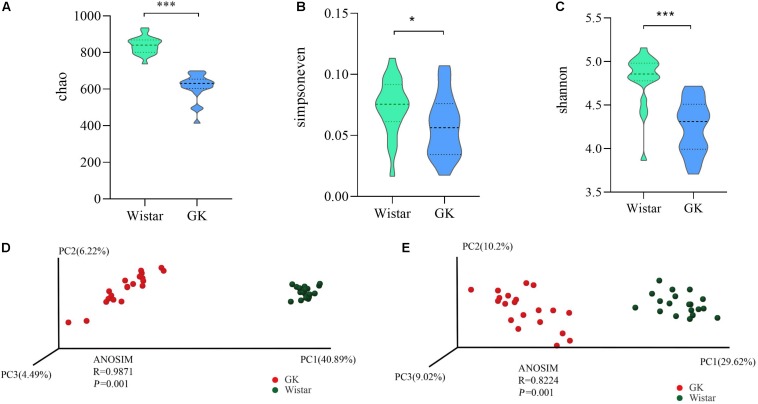
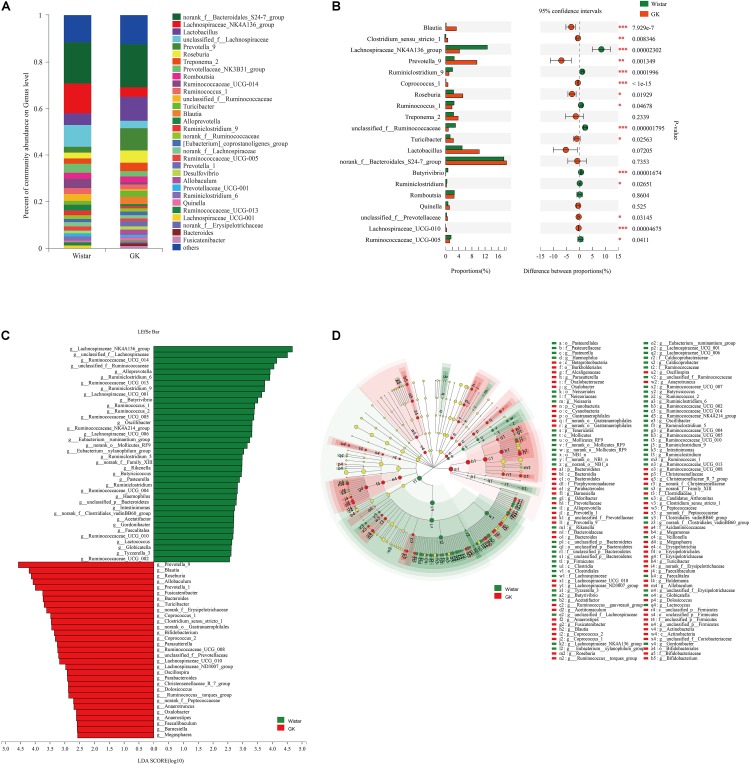
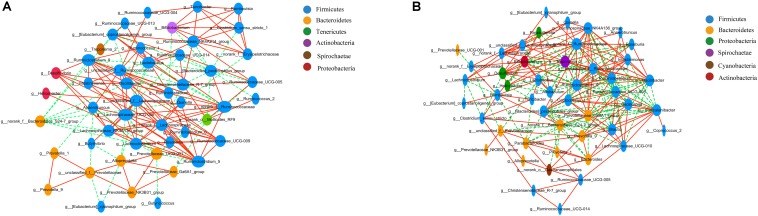
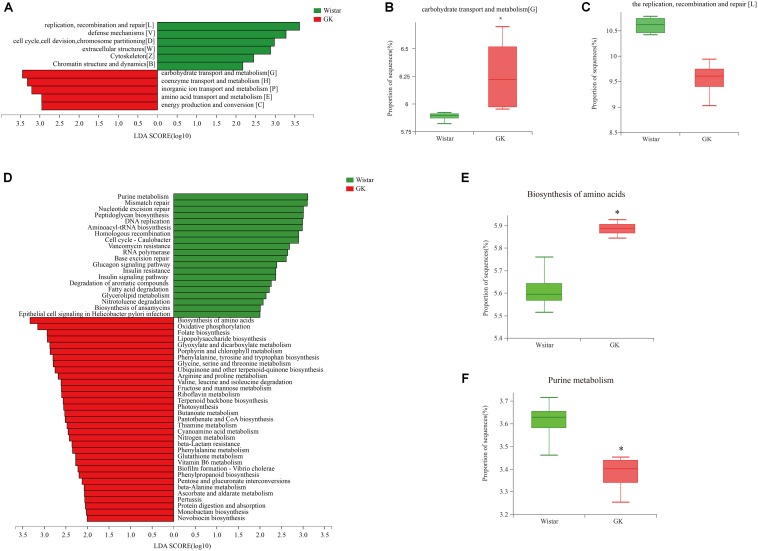
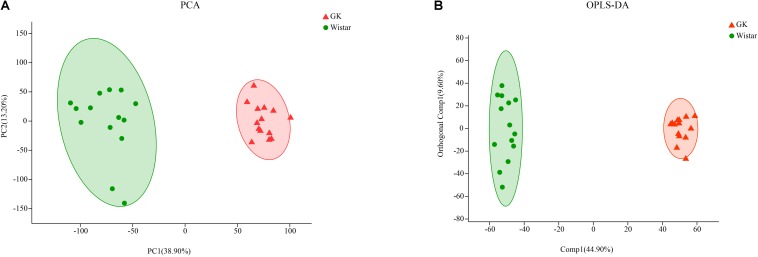
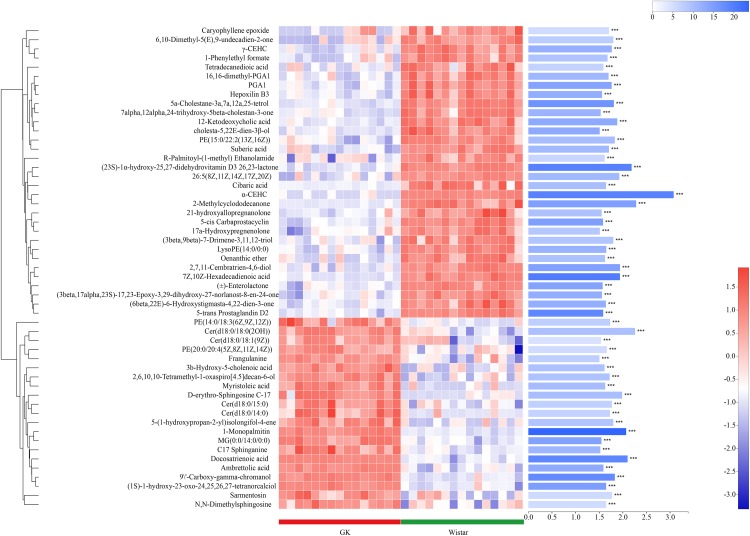
Type 2 diabetes mellitus (T2DM) is one of the most prevalent endocrine diseases in the world. Recent studies have shown that dysbiosis of the gut microbiota may be an important contributor to T2DM pathogenesis. However, the mechanisms underlying the roles of the gut microbiome and fecal metabolome in T2DM have not been characterized. Recently, the Goto-Kakizaki (GK) rat model of T2DM was developed to study the clinical symptoms and characteristics of human T2DM. To further characterize T2DM pathogenesis, we combined multi-omics techniques, including 16S rRNA gene sequencing, metagenomic sequencing, and metabolomics, to analyze gut microbial compositions and functions, and further characterize fecal metabolomic profiles in GK rats. Our results showed that gut microbial compositions were significantly altered in GK rats, as evidenced by reduced microbial diversity, altered microbial taxa distribution, and alterations in the interaction network of the gut microbiome. Functional analysis based on the cluster of orthologous groups (COG) and Kyoto Encyclopedia of Genes and Genomes (KEGG) annotations suggested that 5 functional COG categories belonged to the metabolism cluster and 33 KEGG pathways related to metabolic pathways were significantly enriched in GK rats. Metabolomics profiling identified 53 significantly differentially abundant metabolites in GK rats, including lipids and lipid-like molecules. These lipids were enriched in the glycerophospholipid metabolic pathway. Moreover, functional correlation analysis showed that some altered gut microbiota families, such as and , significantly correlated with alterations in fecal metabolites. Collectively, the results suggested that an altered gut microbiota is associated with T2DM pathogenesis.
2型糖尿病(T2DM)是世界上最常见的内分泌疾病之一。最近的研究表明,肠道微生物群失调可能是T2DM发病机制的一个重要因素。然而,肠道微生物组和粪便代谢组在T2DM中的作用机制尚未明确。最近,为了研究人类T2DM的临床症状和特征,开发了T2DM的Goto-Kakizaki(GK)大鼠模型。为了进一步明确T2DM的发病机制,我们结合了多种组学技术,包括16S rRNA基因测序、宏基因组测序和代谢组学,来分析肠道微生物组成和功能,并进一步明确GK大鼠的粪便代谢组学特征。我们的结果表明,GK大鼠的肠道微生物组成发生了显著变化,表现为微生物多样性降低、微生物分类群分布改变以及肠道微生物组相互作用网络的改变。基于直系同源簇(COG)和京都基因与基因组百科全书(KEGG)注释的功能分析表明,5个功能COG类别属于代谢簇,33条与代谢途径相关的KEGG途径在GK大鼠中显著富集。代谢组学分析确定了GK大鼠中53种显著差异丰富的代谢物,包括脂质和类脂质分子。这些脂质在甘油磷脂代谢途径中富集。此外,功能相关性分析表明,一些改变的肠道微生物群家族,如 和 ,与粪便代谢物的改变显著相关。总的来说,结果表明肠道微生物群的改变与T2DM发病机制有关。