Antwerpen Markus H, Georgi Enrico, Nikolic Alexandra, Zoeller Gudrun, Wohlsein Peter, Baumgärtner Wolfgang, Peyrefitte Christophe, Charrel Remi, Meyer Hermann
Bundeswehr Institute of Microbiology, Munich, Germany.
Small Animal Clinic, University of Veterinary Medicine, Hanover, Germany.
PeerJ. 2019 Mar 1;7:e6561. doi: 10.7717/peerj.6561. eCollection 2019.
Between 2008 and 2011 about 40 cases of human cowpox were reported from Germany and France. Infections had been acquired via close contact to infected, young pet rats. An identical and unique sequence of the hemagglutinin gene was found in various cowpox virus (CPXV) isolates pointing to a common source of infection. In a second CPXV outbreak in cats in a small animal clinic in Germany in 2015, four out of five hospitalized cats showed identical hemagglutinin sequences and thus, a hospital-acquired transmission had been assumed. Next-Generation Sequencing was performed in order to re-investigate the outbreaks, as epidemiological data could not confirm all cases.
Homogenates of lesion material from rats, cats and humans were cultivated in cell culture. The genomes of four virus isolates, nine CPXVs from our strain collections and from DNA of three paraffin-embedded lesion materials were determined by Next Generation Sequencing (NGS). For phylogenetic analyses a MAFFT-alignment was generated. A distance matrix based on concatenated SNPs was calculated and plotted as dendrogram using Unweighted Pair Group Method with Arithmetic mean (UPGMA) for visualization.
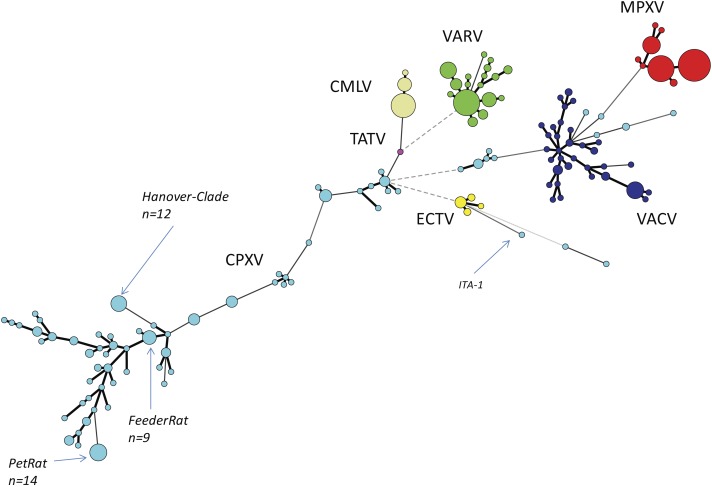
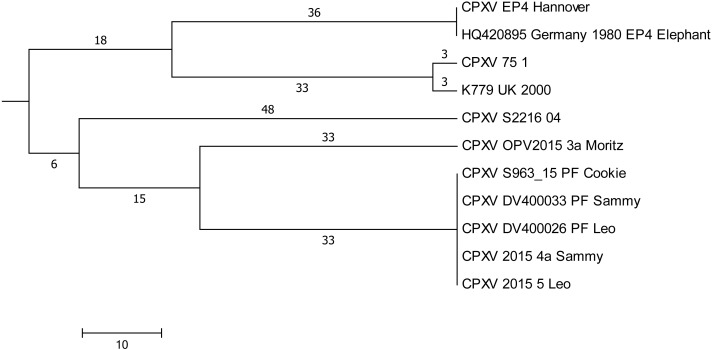
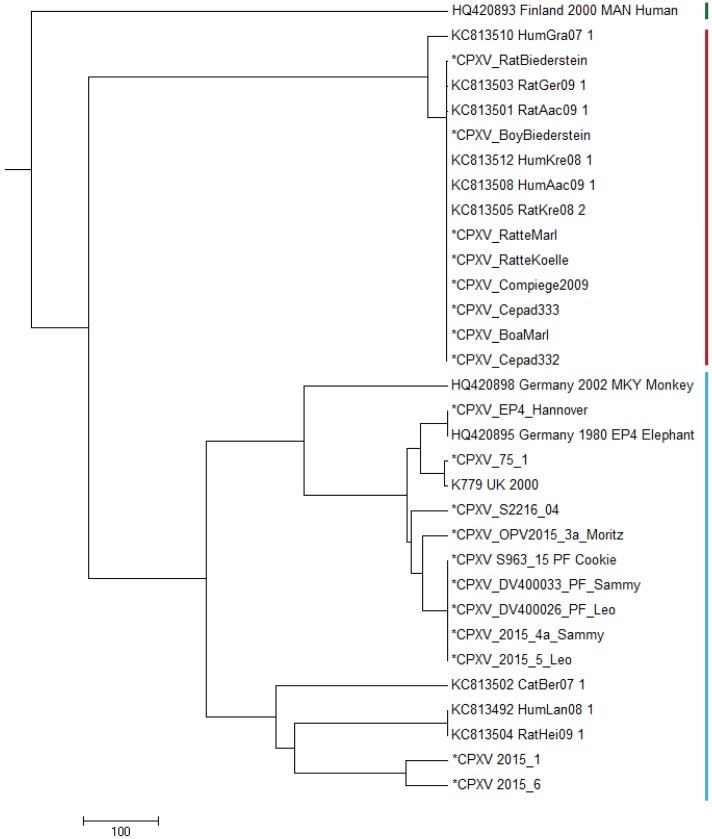
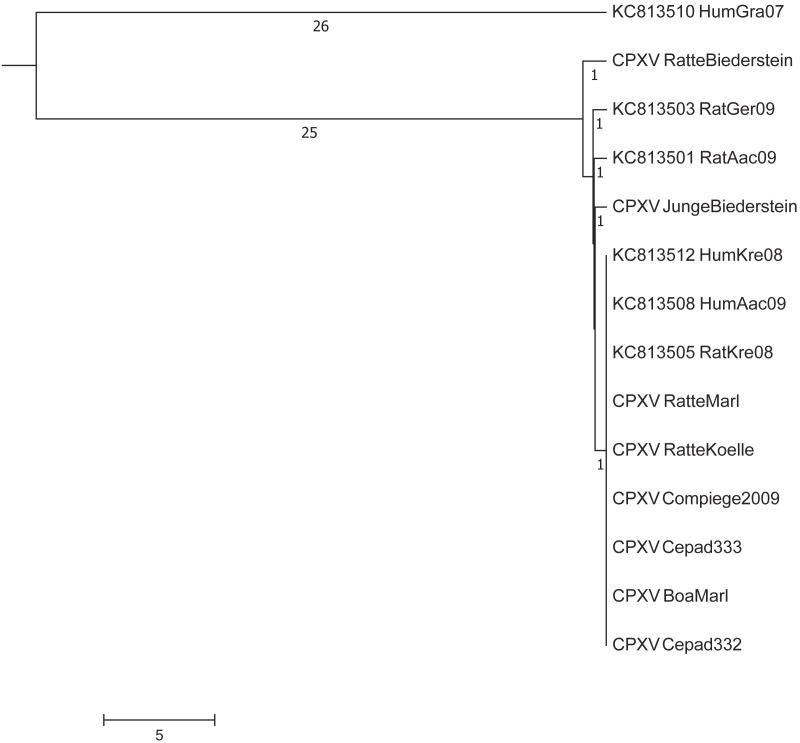
Aligning of about 200.000 nucleotides of 8 virus isolates associated with the pet rat outbreak revealed complete identity of six genomes, the remainder two genomes differed in as little as 3 SNPs. When comparing this dataset with four already published CPXV genomes also associated with the pet rat outbreak, again a maximum difference of 3 SNPs was found. The outbreak which lasted from 2008 till 2011 was indeed caused by a single strain which has maintained an extremely high level of clonality over 4 years. Aligning genomic sequences from four cases of feline cowpox revealed 3 identical sequences and one sequence which differed in 65 nucleotides. Although identical hemagglutinin sequences had been obtained from four hospitalized cats, genomic sequencing proved that a hospital-acquired transmission had occurred in only three cats.
Analyzing the rather short sequence of the hemagglutinin gene is not sufficient to conduct molecular trace back analyses. Instead, whole genome sequencing is the method of choice which can even be applied to paraffin-embedded specimens.
2008年至2011年间,德国和法国报告了约40例人感染牛痘的病例。这些感染是通过与受感染的幼年宠物鼠密切接触而获得的。在各种牛痘病毒(CPXV)分离株中发现了血凝素基因的相同且独特的序列,表明存在共同的感染源。2015年,德国一家小动物诊所发生了猫感染牛痘病毒的第二次疫情,住院的五只猫中有四只血凝素序列相同,因此推测存在医院内传播。由于流行病学数据无法证实所有病例,因此进行了下一代测序以重新调查这些疫情。
将来自大鼠、猫和人类的病变材料匀浆在细胞培养中培养。通过下一代测序(NGS)确定了四个病毒分离株、我们菌株库中的九个CPXV以及三个石蜡包埋病变材料的DNA的基因组。为了进行系统发育分析,生成了MAFFT比对。计算基于串联单核苷酸多态性(SNP)的距离矩阵,并使用算术平均未加权对组方法(UPGMA)绘制为树状图以进行可视化。
对与宠物鼠疫情相关的8个病毒分离株的约200,000个核苷酸进行比对,发现6个基因组完全相同,其余两个基因组仅相差3个SNP。将该数据集与另外四个已发表的同样与宠物鼠疫情相关的CPXV基因组进行比较时,再次发现最大差异为3个SNP。从2008年持续到2011年的疫情确实是由单一菌株引起的,该菌株在4年中保持了极高的克隆性。对四例猫牛痘的基因组序列进行比对,发现3个相同序列和1个相差65个核苷酸的序列。尽管从四只住院猫中获得了相同的血凝素序列,但基因组测序证明只有三只猫发生了医院内传播。
分析血凝素基因的较短序列不足以进行分子溯源分析。相反,全基因组测序是首选方法,甚至可以应用于石蜡包埋的标本。