Department of Medical Biochemistry, MTA-SE Laboratory for Neurobiochemistry, Semmelweis University, 37-47 Tuzolto Street, Budapest, 1094, Hungary.
Neurochem Res. 2019 Oct;44(10):2307-2313. doi: 10.1007/s11064-019-02766-9. Epub 2019 Mar 7.
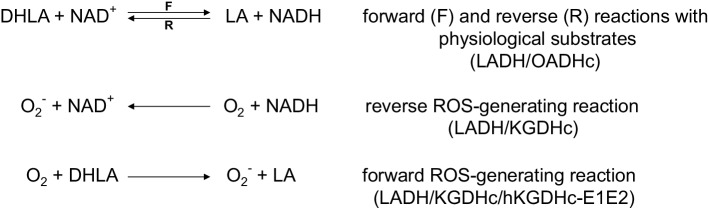
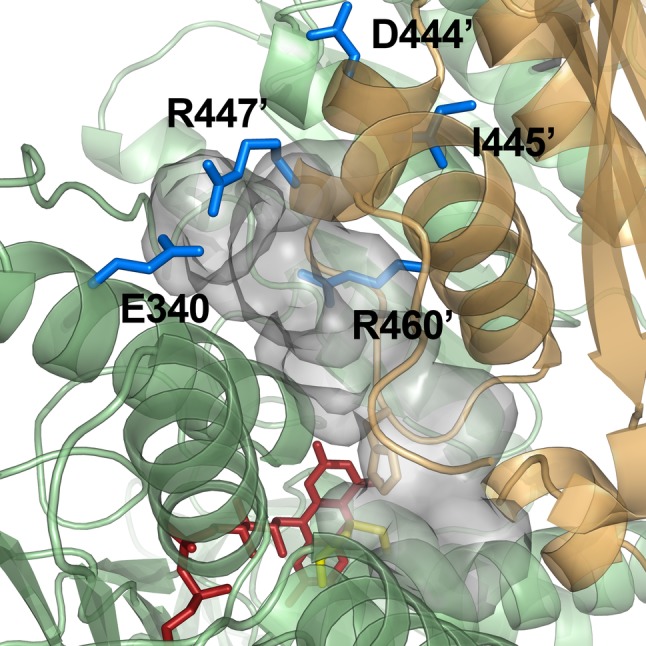
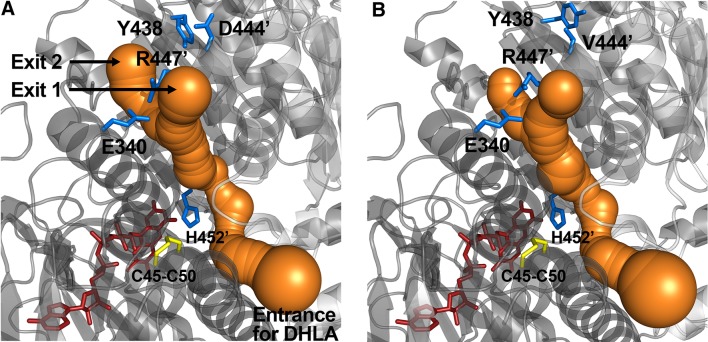
Dihydrolipoamide dehydrogenase (LADH, E3) deficiency is a rare (autosomal, recessive) genetic disorder generally presenting with an onset in the neonatal age and early death; the highest carrier rate has been found among Ashkenazi Jews. Acute clinical episodes usually involve severe metabolic decompensation and lactate acidosis that result in neurological, cardiological, and/or hepatological manifestations. Clinical severity is due to the fact that LADH is a common E3 subunit to the alpha-ketoglutarate, pyruvate, alpha-ketoadipate, and branched-chain alpha-keto acid dehydrogenase complexes, and is also a constituent in the glycine cleavage system, thus a loss in LADH function adversely affects multiple key metabolic routes. However, the severe clinical pictures frequently still do not parallel the LADH activity loss, which implies the involvement of auxiliary biochemical mechanisms; enhanced reactive oxygen species generation as well as affinity loss for multienzyme complexes proved to be key auxiliary exacerbating pathomechanisms. This review provides an overview and an up-to-date molecular insight into the pathomechanisms of this disease in light of the structural conclusions drawn from the first crystal structure of a disease-causing hE3 variant determined recently in our laboratory.
二氢硫辛酰胺脱氢酶(LADH,E3)缺乏症是一种罕见的(常染色体隐性)遗传疾病,通常在新生儿期发病并导致早期死亡;在阿什肯纳兹犹太人中发现了最高的携带率。急性临床发作通常涉及严重的代谢失代偿和乳酸酸中毒,导致神经、心脏和/或肝脏表现。临床严重程度是由于 LADH 是 α-酮戊二酸、丙酮酸、α-酮戊二酸、支链α-酮酸脱氢酶复合物的常见 E3 亚基,也是甘氨酸裂解系统的组成部分,因此 LADH 功能的丧失会对多个关键代谢途径产生不利影响。然而,严重的临床症状通常仍与 LADH 活性丧失不一致,这表明辅助生化机制的参与;增强的活性氧生成以及对多酶复合物的亲和力丧失已被证明是关键的辅助加重发病机制。鉴于最近我们实验室首次确定的致病 hE3 变异体的晶体结构得出的结构结论,本综述提供了对该疾病发病机制的概述和最新的分子见解。