Centre de Bioinformatique, Biostatistique et Biologie Intégrative (C3BI), Institut Pasteur, Paris, France.
Department of Epidemiology, Harvard T.H. Chan School of Public Health, Boston, Massachusetts, United States of America.
PLoS Genet. 2019 Mar 8;15(3):e1008018. doi: 10.1371/journal.pgen.1008018. eCollection 2019 Mar.
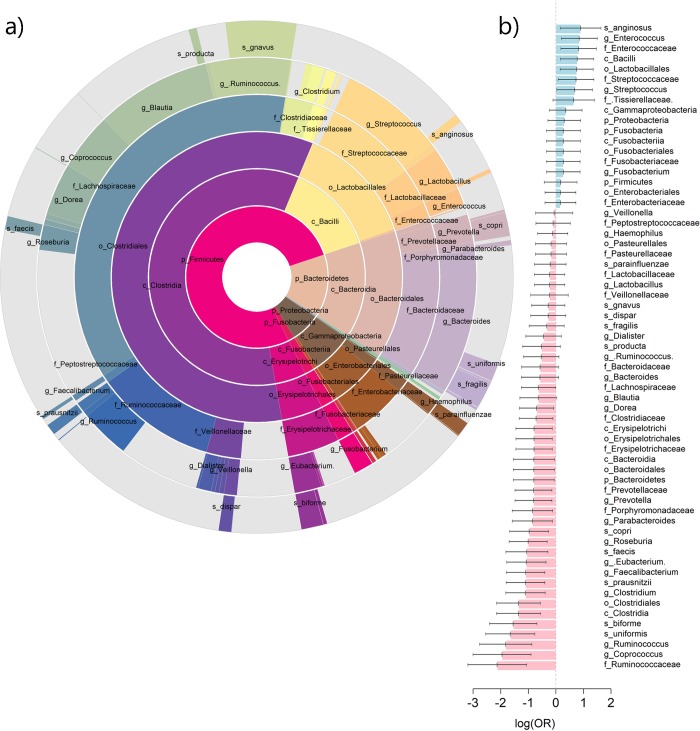
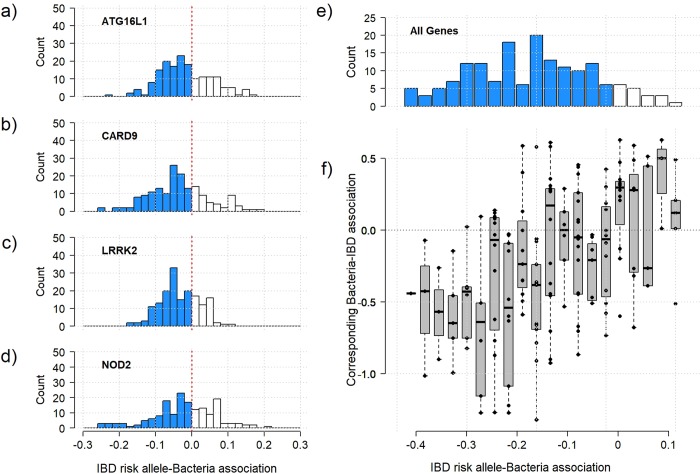
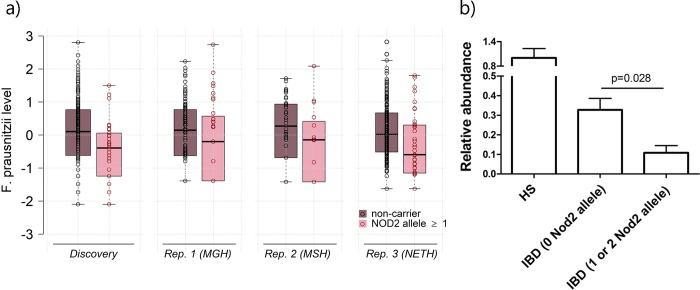
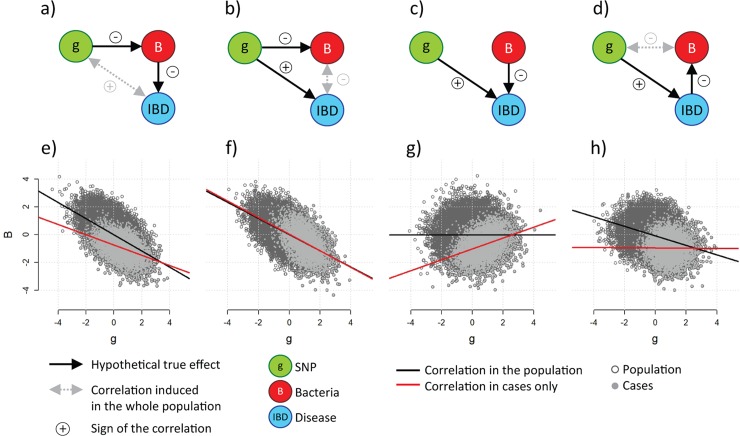
Several bacteria in the gut microbiota have been shown to be associated with inflammatory bowel disease (IBD), and dozens of IBD genetic variants have been identified in genome-wide association studies. However, the role of the microbiota in the etiology of IBD in terms of host genetic susceptibility remains unclear. Here, we studied the association between four major genetic variants associated with an increased risk of IBD and bacterial taxa in up to 633 IBD cases. We performed systematic screening for associations, identifying and replicating associations between NOD2 variants and two taxa: the Roseburia genus and the Faecalibacterium prausnitzii species. By exploring the overall association patterns between genes and bacteria, we found that IBD risk alleles were significantly enriched for associations concordant with bacteria-IBD associations. To understand the significance of this pattern in terms of the study design and known effects from the literature, we used counterfactual principles to assess the fitness of a few parsimonious gene-bacteria-IBD causal models. Our analyses showed evidence that the disease risk of these genetic variants were likely to be partially mediated by the microbiome. We confirmed these results in extensive simulation studies and sensitivity analyses using the association between NOD2 and F. prausnitzii as a case study.
肠道微生物群中的几种细菌已被证明与炎症性肠病(IBD)有关,并且在全基因组关联研究中已经确定了数十种 IBD 遗传变异。然而,微生物群在宿主遗传易感性方面在 IBD 的发病机制中的作用仍不清楚。在这里,我们研究了与 IBD 风险增加相关的四个主要遗传变异与多达 633 例 IBD 病例中的细菌分类群之间的关联。我们进行了系统的关联筛选,鉴定并复制了 NOD2 变异与两种分类群之间的关联:Roseburia 属和 Faecalibacterium prausnitzii 种。通过探索基因与细菌之间的整体关联模式,我们发现 IBD 风险等位基因与与细菌-IBD 关联一致的关联显著富集。为了理解这种模式在研究设计和文献中已知影响方面的意义,我们使用反事实原则来评估一些简单的基因-细菌-IBD 因果模型的适应性。我们的分析表明,这些遗传变异的疾病风险很可能部分是由微生物组介导的。我们使用 NOD2 和 F. prausnitzii 之间的关联作为案例研究,在广泛的模拟研究和敏感性分析中证实了这些结果。