Arfianti Evi, Larter Claire Z, Lee Seungsoo, Barn Vanessa, Haigh Geoffrey, Yeh Matthew M, Ioannou George N, Teoh Narci C, Farrell Geoffrey C
Liver Research Group, Australian National University Medical School, The Canberra Hospital, Australian Capital Territory, Australia.
Division of Gastroenterology, University of Washington, Seattle, Washington, United States.
J Clin Transl Res. 2016 Jan 19;2(1):26-37. eCollection 2016 Apr 15.
There are strong links between obesity, diabetes and hepatocellular carcinoma (HCC), but molecular mechanisms remain unclear.
We tested the proposed involvement of NF-κB, IL-6/STAT3 and Akt/mTORC1 before onset (at 3 months) and at onset (6 months) of accelerated hepatocarcinogenesis in DEN-injected obese and diabetic compared to lean wildtype () mice, and also studied the hepatocyte proliferative response to DNA damage between the obese and lean lines.
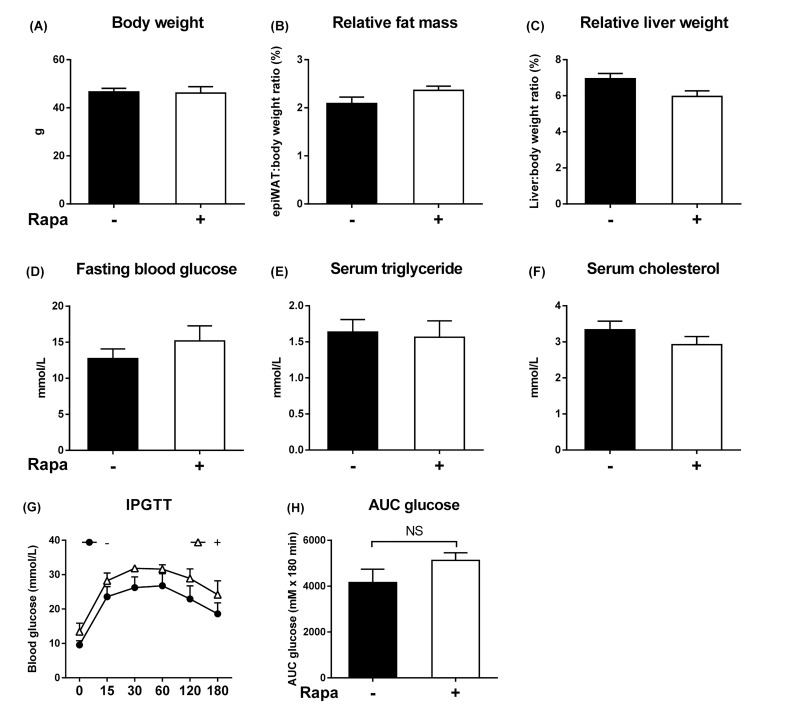
Male and littermates fed normal chow were DEN-injected (10mg/kg i.p.) at age 12-15 days. To test the effect of mTOR inhibitor on growth of dysplastic hepatocytes, a separate cohort of DEN-injected mice was administered rapamycin (4 mg/kg body weight/day).
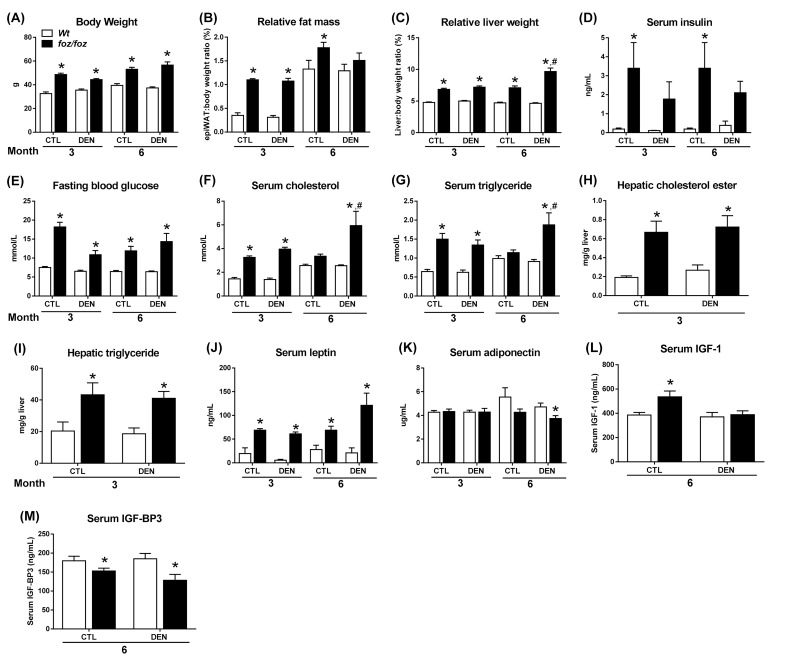
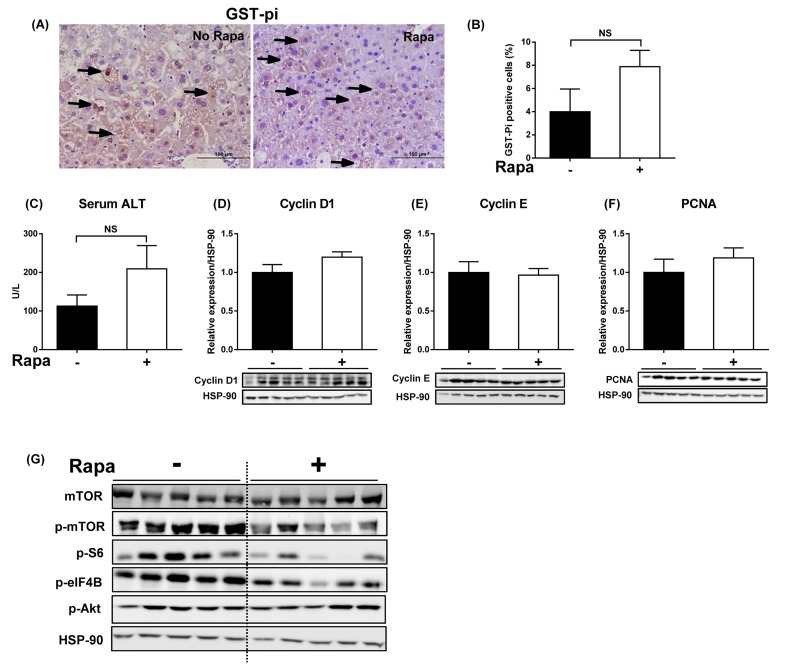
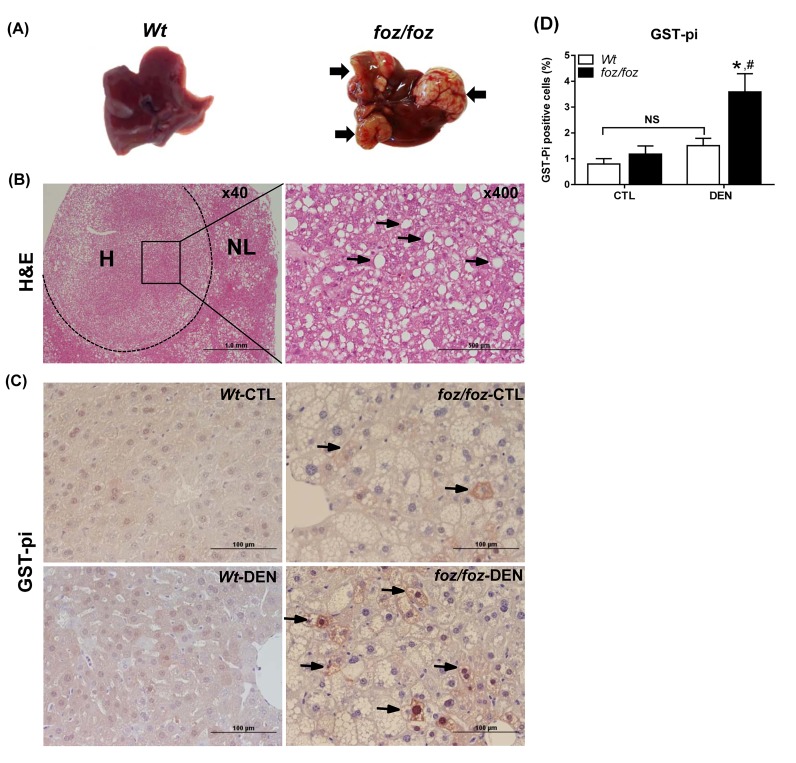
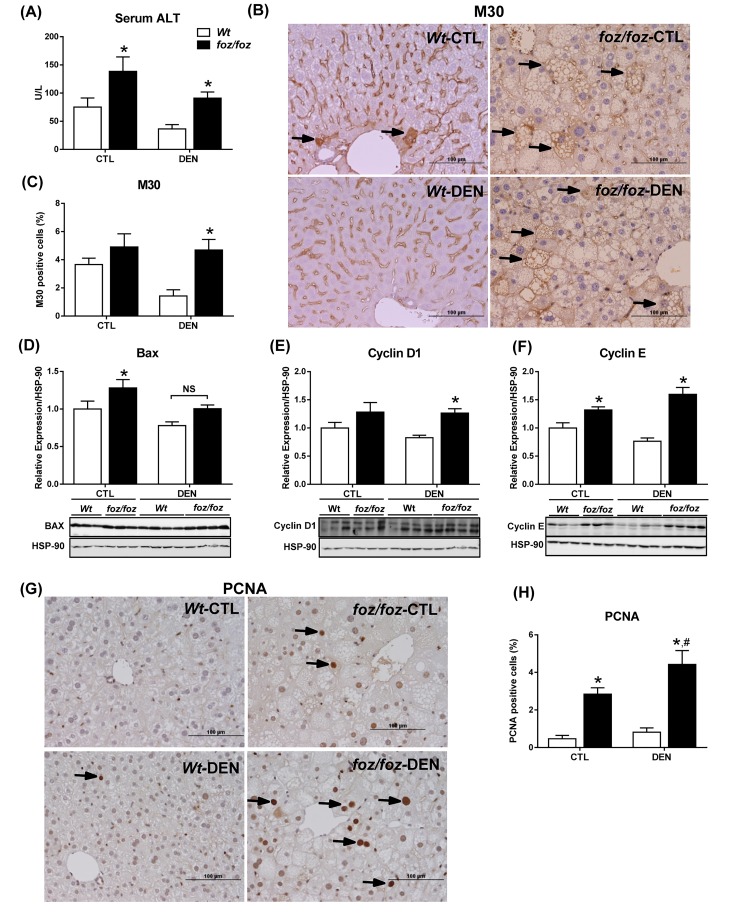
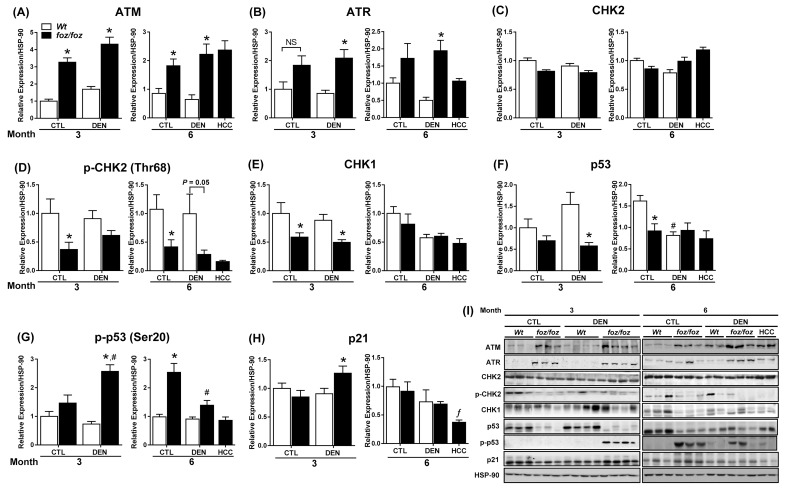
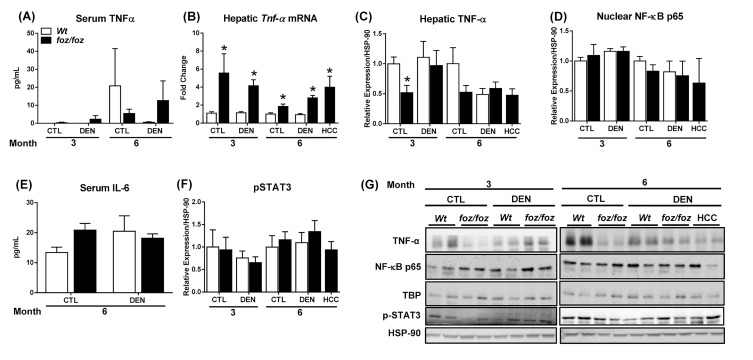
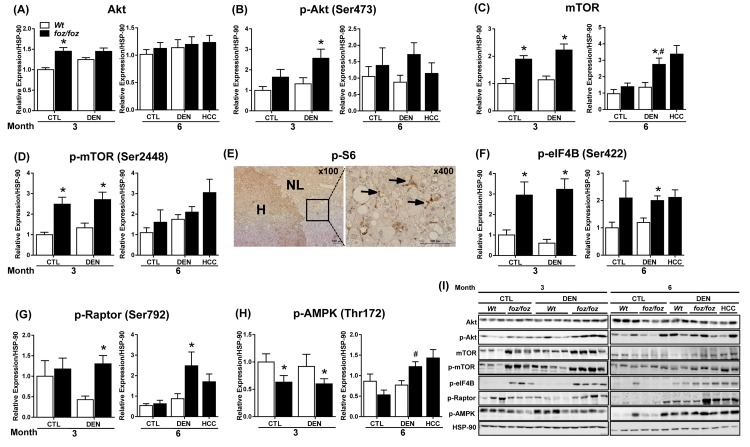
mice developed obesity, hyperinsulinemia, diabetes, adipokine dysregulation and fatty liver, without increased serum or liver TNF-α or serum IL-6. All DEN-injected mice developed HCC by 6 mths . 0/10 lean . At 3 mths, there were more dysplastic hepatocytes in DEN-injected than , with increased liver injury (serum ALT), hepatocyte apoptosis (M30-positive cells) and proliferation (cyclin D1, cyclin E, PCNA), but neither NF-κB nor STAT3 activation. livers exhibited upregulation of DNA damage sensors ATM and ATR, with inadequate cell cycle checkpoint controls (CHK1, CHK2, p53, p21). Akt and mTORC1 were highly activated in livers from mice. Despite such activation, rapamycin failed to reduce growth of dysplastic hepatocytes.
Accelerated DEN-induced HCC in obese/diabetic mice is linked to enhanced growth of dysplastic hepatocytes that cannot be attributed to NF-κB or IL-6/STAT3 activation, nor to sustained mTORC1 activation. The critical mechanism for obesity-enhanced hepatocarcinogenesis lies in the disconnection between hepatocellular injury with DNA damage, and an unrestrained proliferative response.
This study supports the epidemiological data linking obesity, diabetes and fatty liver disease with increased risk for developing HCC. The findings also suggest that mTORC1 inhibition may not be beneficial in the prevention of obesity-related hepatocarcinogenesis.
肥胖、糖尿病与肝细胞癌(HCC)之间存在紧密联系,但分子机制仍不清楚。
我们检测了在二乙基亚硝胺(DEN)注射的肥胖和糖尿病小鼠与瘦的野生型小鼠相比,在加速肝癌发生前(3个月时)和发生时(6个月时),核因子κB(NF-κB)、白细胞介素-6/信号转导和转录激活因子3(IL-6/STAT3)以及蛋白激酶B(Akt)/哺乳动物雷帕霉素靶蛋白复合物1(mTORC1)是否如所提出的那样参与其中,并且还研究了肥胖和瘦品系之间肝细胞对DNA损伤的增殖反应。
12 - 15日龄的雄性野生型和同窝小鼠喂食正常饲料,腹腔注射DEN(10mg/kg)。为了测试mTOR抑制剂对发育异常肝细胞生长的影响,另一组腹腔注射DEN的野生型小鼠给予雷帕霉素(4mg/kg体重/天)。
野生型小鼠出现肥胖、高胰岛素血症、糖尿病、脂肪因子失调和脂肪肝,血清或肝脏肿瘤坏死因子-α(TNF-α)或血清白细胞介素-6未增加。所有腹腔注射DEN的野生型小鼠在6个月时都发生了HCC。10只瘦小鼠中0只发生。在3个月时,腹腔注射DEN的野生型小鼠中发育异常的肝细胞比瘦小鼠更多,伴有肝损伤增加(血清丙氨酸氨基转移酶[ALT])、肝细胞凋亡(M30阳性细胞)和增殖(细胞周期蛋白D1、细胞周期蛋白E、增殖细胞核抗原[PCNA])增加,但NF-κB和STAT3均未激活。野生型小鼠肝脏中DNA损伤传感器共济失调毛细血管扩张突变蛋白(ATM)和毛细血管扩张性共济失调和Rad3相关蛋白(ATR)上调,细胞周期检查点控制不足(细胞周期检查点激酶1[CHK1]、细胞周期检查点激酶2[CHK2]、p53蛋白(polyomavirus tumor antigen)、p21蛋白)。Akt和mTORC1在野生型小鼠肝脏中高度激活。尽管有这种激活,雷帕霉素未能降低发育异常肝细胞的生长。
肥胖/糖尿病小鼠中DEN诱导的HCC加速与发育异常肝细胞的生长增强有关,这不能归因于NF-κB或IL-6/STAT3激活,也不能归因于mTORC1的持续激活。肥胖增强肝癌发生的关键机制在于肝细胞损伤伴DNA损伤与无节制的增殖反应之间的脱节。
本研究支持将肥胖、糖尿病和脂肪肝病与发生HCC风险增加联系起来的流行病学数据。研究结果还表明,抑制mTORC1可能对预防肥胖相关肝癌发生无益。