Institute for Animal Breeding and Genetics, University of Veterinary Medicine Hannover, Bünteweg 17p, Hannover, 30559, Germany.
Research Center for Emerging Infections and Zoonoses, University of Veterinary Medicine Hannover, Bünteweg 17p, Hannover, 30559, Germany.
BMC Bioinformatics. 2019 Mar 15;20(1):144. doi: 10.1186/s12859-019-2705-9.
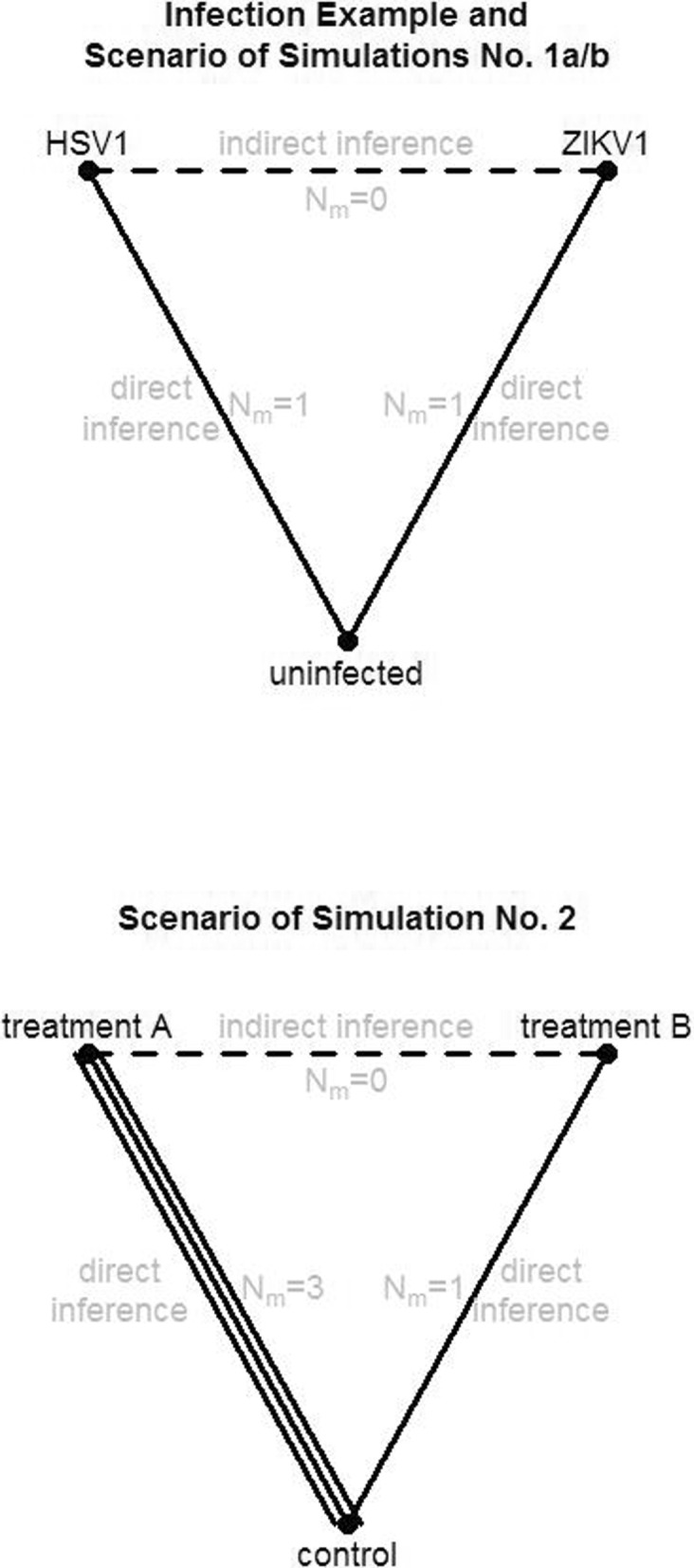
Using meta-analysis, high-dimensional transcriptome expression data from public repositories can be merged to make group comparisons that have not been considered in the original studies. Merging of high-dimensional expression data can, however, implicate batch effects that are sometimes difficult to be removed. Removing batch effects becomes even more difficult when expression data was taken using different technologies in the individual studies (e.g. merging of microarray and RNA-seq data). Network meta-analysis has so far not been considered to make indirect comparisons in transcriptome expression data, when data merging appears to yield biased results.
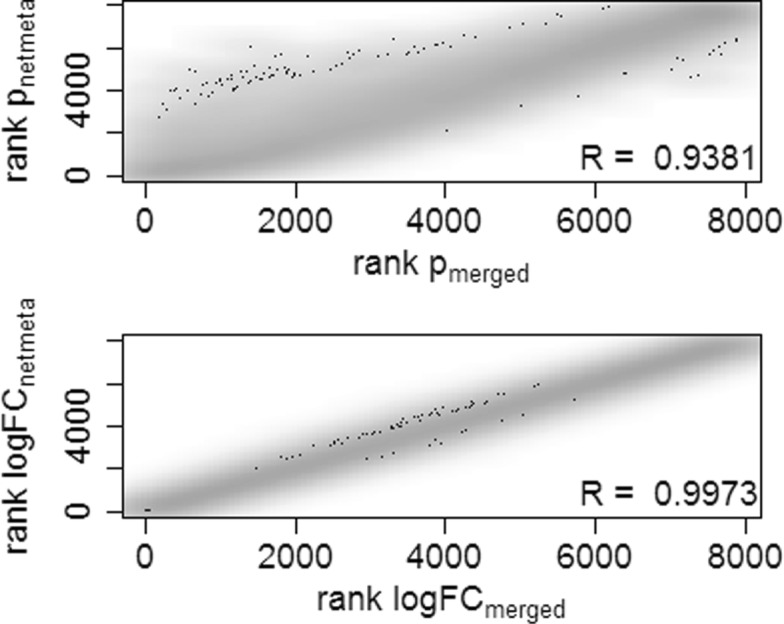
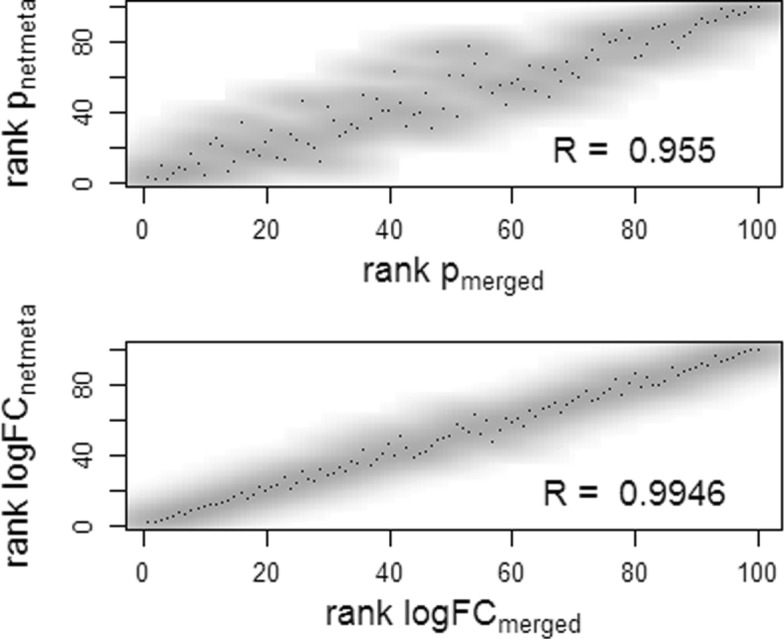
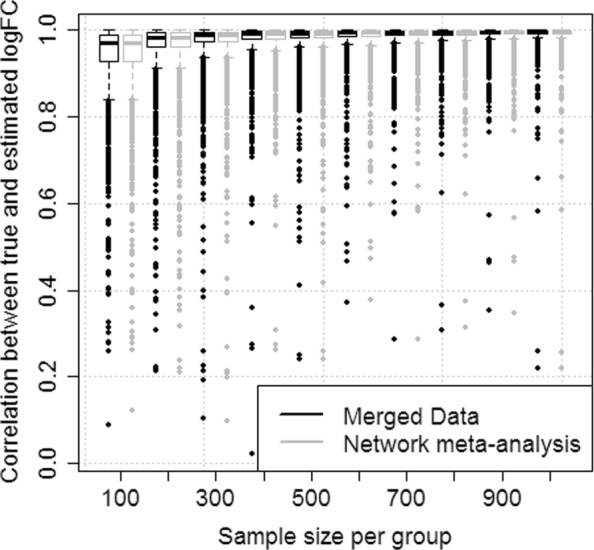
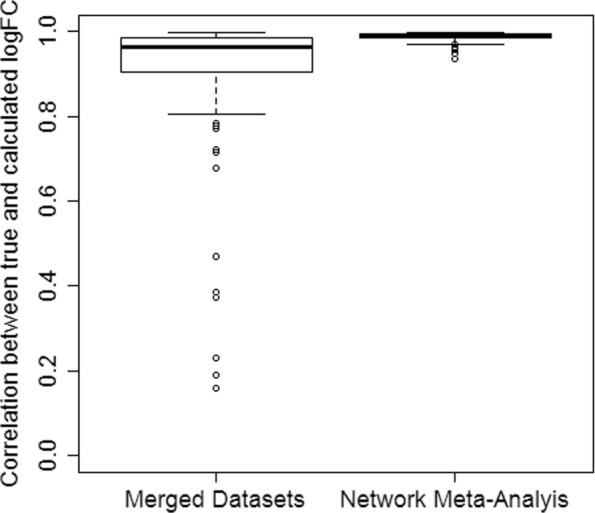
We demonstrate in a simulation study that the results from analyzing merged data sets and the results from network meta-analysis are highly correlated in simple study networks. In the case that an edge in the network is supported by multiple independent studies, network meta-analysis produces fold changes that are closer to the simulated ones than those obtained from analyzing merged data sets. Finally, we also demonstrate the practicability of network meta-analysis on a real-world data example from neuroinfection research.
Network meta-analysis is a useful means to make new inferences when combining multiple independent studies of molecular, high-throughput expression data. This method is especially advantageous when batch effects between studies are hard to get removed.
利用荟萃分析,可以合并来自公共存储库的高维转录组表达数据,以进行原始研究中未考虑的组间比较。然而,高维表达数据的合并可能会涉及到批次效应,而这些效应有时很难消除。当个体研究中使用不同的技术(例如,微阵列和 RNA-seq 数据的合并)获取表达数据时,去除批次效应就变得更加困难。到目前为止,网络荟萃分析尚未被认为可用于在转录组表达数据中进行间接比较,因为数据合并似乎会产生有偏的结果。
我们在一项模拟研究中证明,在简单的研究网络中,分析合并数据集的结果和网络荟萃分析的结果高度相关。在网络中的边由多个独立的研究支持的情况下,网络荟萃分析产生的倍数变化比从分析合并数据集得到的结果更接近模拟结果。最后,我们还在神经感染研究的真实世界数据示例中展示了网络荟萃分析的实用性。
当组合多个独立的分子、高通量表达数据研究时,网络荟萃分析是一种进行新推断的有用方法。当研究之间的批次效应难以消除时,这种方法尤其有利。