Paul Bindu D, Snyder Solomon H
The Solomon H. Snyder Department of Neuroscience, Johns Hopkins University School of Medicine, Baltimore, MD, United States.
Department of Psychiatry and Behavioral Sciences, Johns Hopkins University School of Medicine, Baltimore, MD, United States.
Front Mol Neurosci. 2019 Mar 19;12:68. doi: 10.3389/fnmol.2019.00068. eCollection 2019.
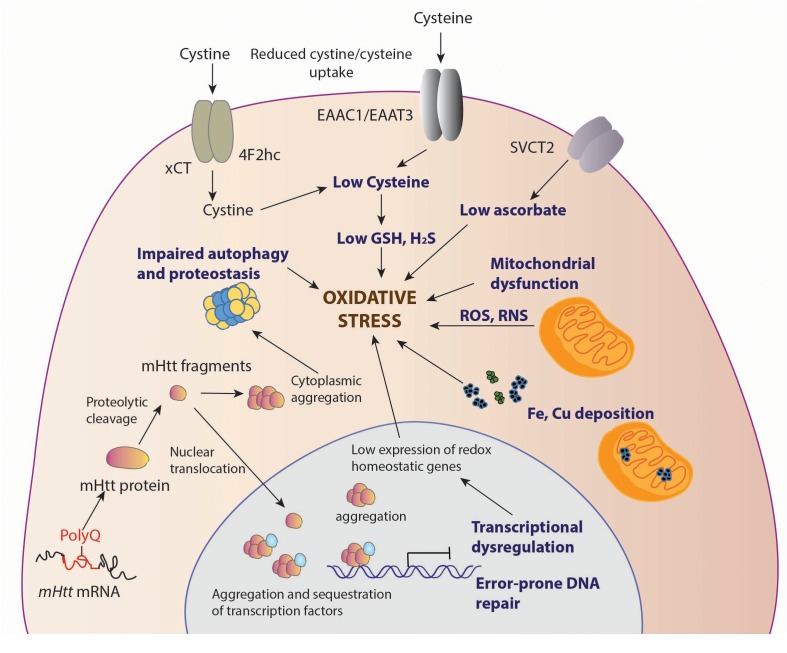
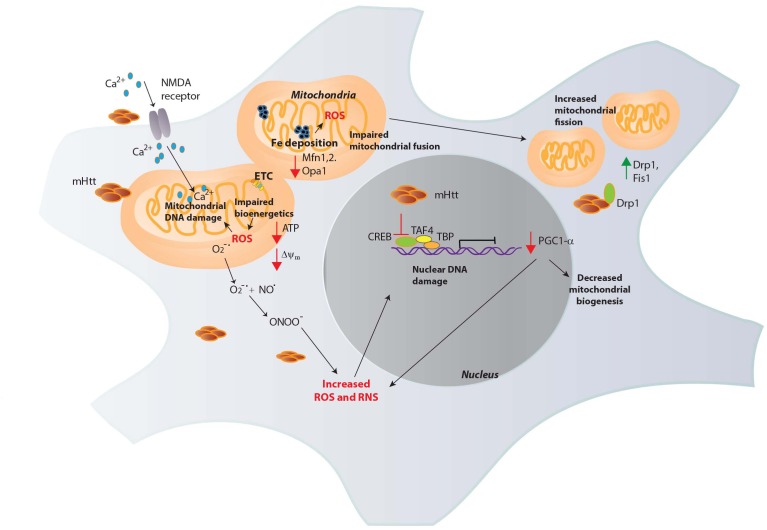
Huntington's disease (HD) is a neurodegenerative disease triggered by expansion of polyglutamine repeats in the protein huntingtin. Mutant huntingtin (mHtt) aggregates and elicits toxicity by multiple mechanisms which range from dysregulated transcription to disturbances in several metabolic pathways in both the brain and peripheral tissues. Hallmarks of HD include elevated oxidative stress and imbalanced redox signaling. Disruption of antioxidant defense mechanisms, involving antioxidant molecules and enzymes involved in scavenging or reversing oxidative damage, have been linked to the pathophysiology of HD. In addition, mitochondrial function is compromised in HD leading to impaired bioenergetics and elevated production of free radicals in cells. However, the exact mechanisms linking redox imbalance to neurodegeneration are still elusive. This review will focus on the current understanding of aberrant redox homeostasis in HD and potential therapeutic interventions.
亨廷顿舞蹈症(HD)是一种神经退行性疾病,由亨廷顿蛋白中多聚谷氨酰胺重复序列的扩增引发。突变型亨廷顿蛋白(mHtt)聚集并通过多种机制引发毒性,这些机制从转录失调到大脑和外周组织中多种代谢途径的紊乱不等。HD的特征包括氧化应激升高和氧化还原信号失衡。抗氧化防御机制的破坏,涉及清除或逆转氧化损伤的抗氧化分子和酶,已与HD的病理生理学相关联。此外,HD中线粒体功能受损,导致生物能量学受损和细胞内自由基产生增加。然而,将氧化还原失衡与神经退行性变联系起来的确切机制仍然难以捉摸。本综述将聚焦于目前对HD中异常氧化还原稳态的理解以及潜在的治疗干预措施。