Department of Chemistry, Boston University, Boston, MA, 02215, USA.
Department of Biomedical Engineering, Boston University, Boston, MA, 02215, USA.
Sci Rep. 2019 Apr 16;9(1):6180. doi: 10.1038/s41598-019-42618-8.
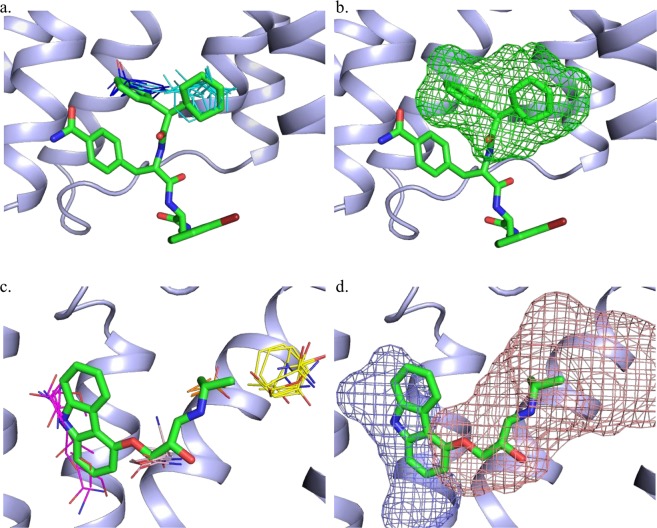
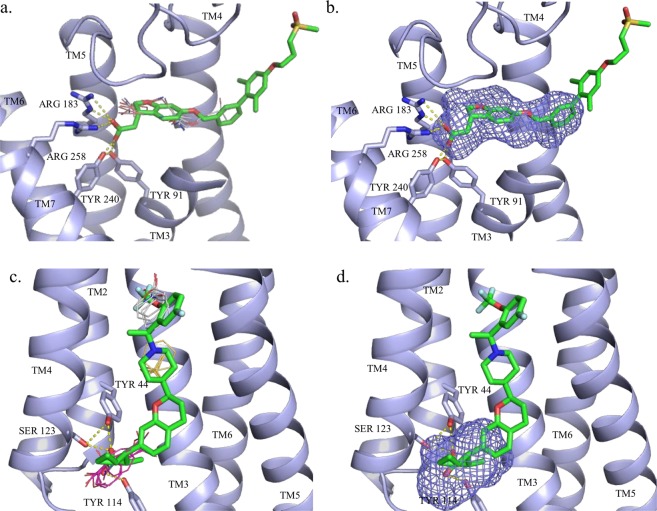
Allosteric modulation of G protein-coupled receptors represent a promising mechanism of pharmacological intervention. Dramatic developments witnessed in the structural biology of membrane proteins continue to reveal that the binding sites of allosteric modulators are widely distributed, including along protein surfaces. Here we restrict consideration to intrahelical and intracellular sites together with allosteric conformational locks, and show that the protein mapping tools FTMap and FTSite identify 83% and 88% of such experimentally confirmed allosteric sites within the three strongest sites found. The methods were also able to find partially hidden allosteric sites that were not fully formed in X-ray structures crystallized in the absence of allosteric ligands. These results confirm that the intrahelical sites capable of binding druglike allosteric modulators are among the strongest ligand recognition sites in a large fraction of GPCRs and suggest that both FTMap and FTSite are useful tools for identifying allosteric sites and to aid in the design of such compounds in a range of GPCR targets.
变构调节 G 蛋白偶联受体代表了一种有前途的药物干预机制。膜蛋白结构生物学的显著发展继续表明,变构调节剂的结合位点广泛分布,包括在蛋白质表面。在这里,我们将研究限制在螺旋内和细胞内位点以及变构构象锁内,并表明蛋白图谱工具 FTMap 和 FTSite 在三个最强的位点中识别出 83%和 88%的此类经实验证实的变构位点。这些方法还能够找到部分隐藏的变构位点,这些位点在没有变构配体的情况下结晶的 X 射线结构中没有完全形成。这些结果证实,能够结合类药物变构调节剂的螺旋内位点是 GPCR 中很大一部分具有最强配体识别位点的位点之一,并表明 FTMap 和 FTSite 都是识别变构位点和辅助此类化合物设计的有用工具,适用于一系列 GPCR 靶标。