Department of Neurology University of Massachusetts Medical School Worcester Massachusetts.
Department of Human Genetics Emory University School of Medicine Atlanta Georgia.
Ann Clin Transl Neurol. 2019 Mar 3;6(4):642-654. doi: 10.1002/acn3.738. eCollection 2019 Apr.
Dysferlin is a large transmembrane protein that functions in critical processes of membrane repair and vesicle fusion. Dysferlin-deficiency due to mutations in the dysferlin gene leads to muscular dystrophy (Miyoshi myopathy (MM), limb girdle muscular dystrophy type 2B (LGMD2B), distal myopathy with anterior tibial onset (DMAT)), typically with early adult onset. At least 416 pathogenic dysferlin mutations are known, but for approximately 17% of patients, one or both of their pathogenic variants remain undefined following standard exon sequencing methods that interrogate exons and nearby flanking intronic regions but not the majority of intronic regions.
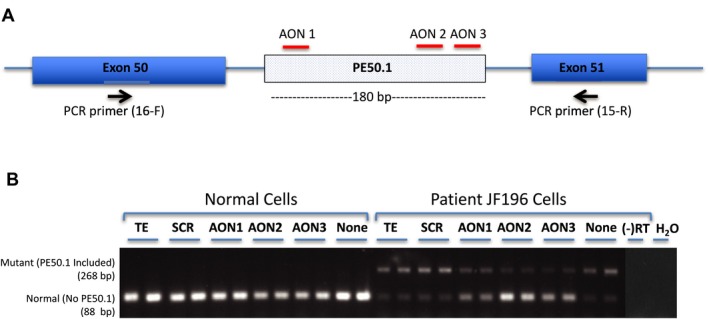
We sequenced RNA from myogenic cells to identify a novel dysferlin pathogenic variant in two affected siblings that previously had only one disease-causing variant identified. We designed antisense oligonucleotides (AONs) to bypass the effects of this mutation on RNA splicing.
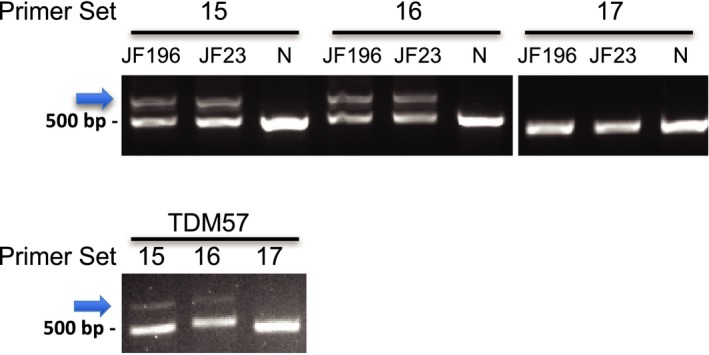
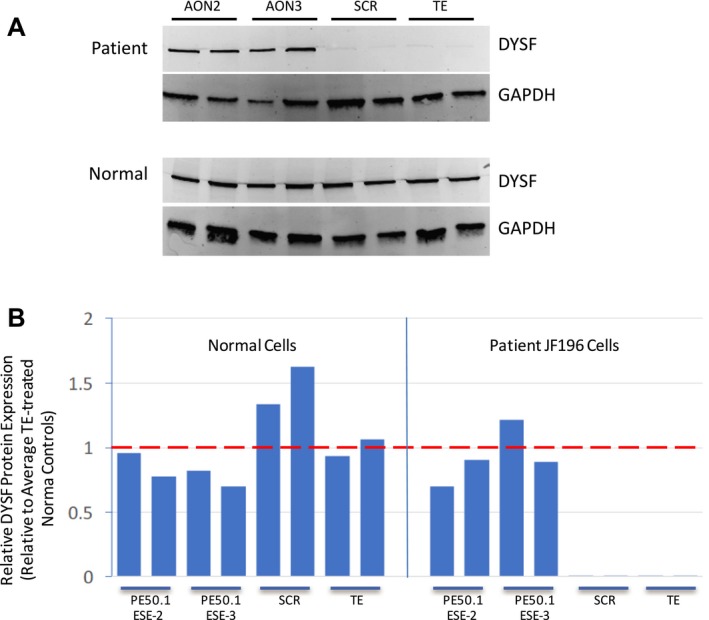
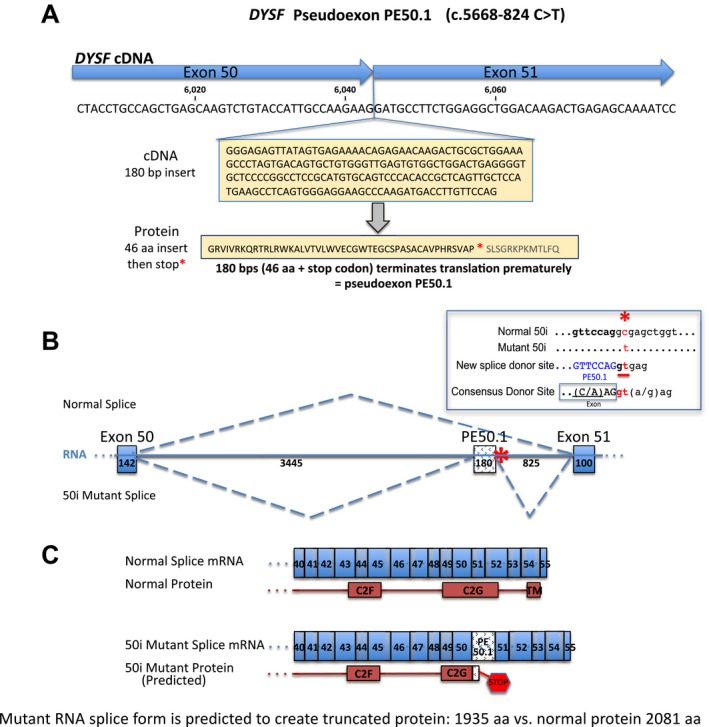
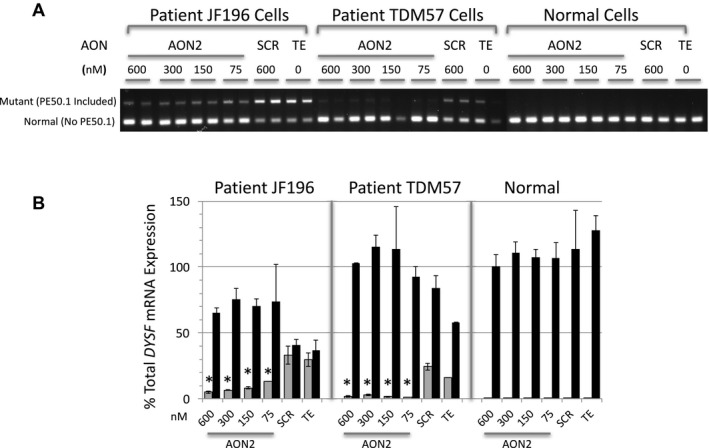
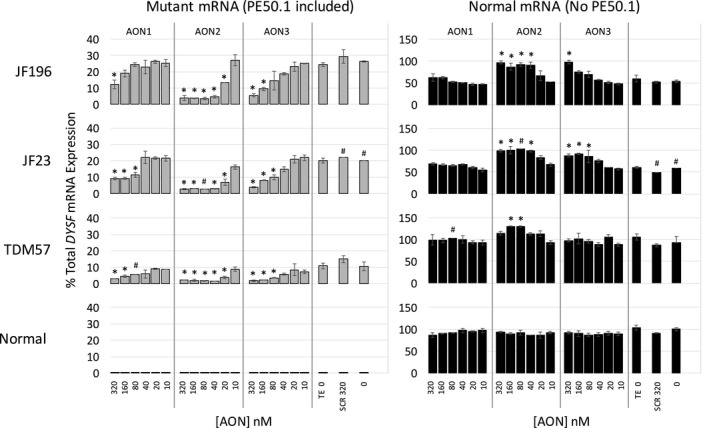
We identified a new pathogenic point mutation deep within dysferlin intron 50i. This intronic variant causes aberrant mRNA splicing and inclusion of an additional pseudoexon (PE, we term PE50.1) within the mature dysferlin mRNA. PE50.1 inclusion alters the protein sequence, causing premature translation termination. We identified this mutation in 23 dysferlinopathy patients (seventeen families), revealing it to be one of the more prevalent dysferlin mutations. We used AON-mediated exon skipping to correct the aberrant PE50.1 splicing events in vitro, which increased normal mRNA production and significantly restored dysferlin protein expression.
Deep intronic mutations can be a common underlying cause of dysferlinopathy, and importantly, could be treatable with AON-based exon-skipping strategies.
肌营养不良蛋白是一种大型跨膜蛋白,在膜修复和囊泡融合的关键过程中发挥作用。由于肌营养不良蛋白基因中的突变导致肌营养不良蛋白缺失,从而导致肌肉疾病(Miyoshi 肌病(MM)、肢带型肌营养不良 2B 型(LGMD2B)、胫骨前肌起始的远端肌病(DMAT)),通常在成年早期发病。已知至少有 416 种致病性肌营养不良蛋白突变,但对于大约 17%的患者,在使用标准外显子测序方法(检测外显子和附近的侧翼内含子区域,但不检测大多数内含子区域)对其致病性变异进行检测后,仍然无法确定一个或两个致病性变异。
我们从肌源性细胞中测序 RNA,以鉴定两个受影响的同胞中一种新的肌营养不良蛋白致病性变异,他们之前仅鉴定出一种致病变异。我们设计了反义寡核苷酸(AON)来绕过该突变对 RNA 剪接的影响。
我们在肌营养不良蛋白内含子 50i 中发现了一个新的致病性点突变。该内含子变体导致异常的 mRNA 剪接,并在成熟的肌营养不良蛋白 mRNA 中包含一个额外的假外显子(PE,我们称之为 PE50.1)。PE50.1 的包含改变了蛋白质序列,导致过早的翻译终止。我们在 23 名肌营养不良蛋白病患者(十七个家庭)中发现了这种突变,表明它是最常见的肌营养不良蛋白突变之一。我们使用 AON 介导的外显子跳跃在体外纠正异常的 PE50.1 剪接事件,这增加了正常 mRNA 的产生,并显著恢复了肌营养不良蛋白的表达。
深内含子突变可能是肌营养不良蛋白病的常见原因,重要的是,可以通过基于 AON 的外显子跳跃策略进行治疗。