Zieman A G, Coulombe P A
Department of Cell and Developmental Biology, University of Michigan Medical School, 3071 Biomedical Sciences Research Building, 109 Zina Pitcher Place, Ann Arbor, MI, 48109, U.S.A.
Department of Dermatology, University of Michigan Medical School, 3071 Biomedical Sciences Research Building, 109 Zina Pitcher Place, Ann Arbor, MI, 48109, U.S.A.
Br J Dermatol. 2020 Mar;182(3):564-573. doi: 10.1111/bjd.18033. Epub 2019 Jul 24.
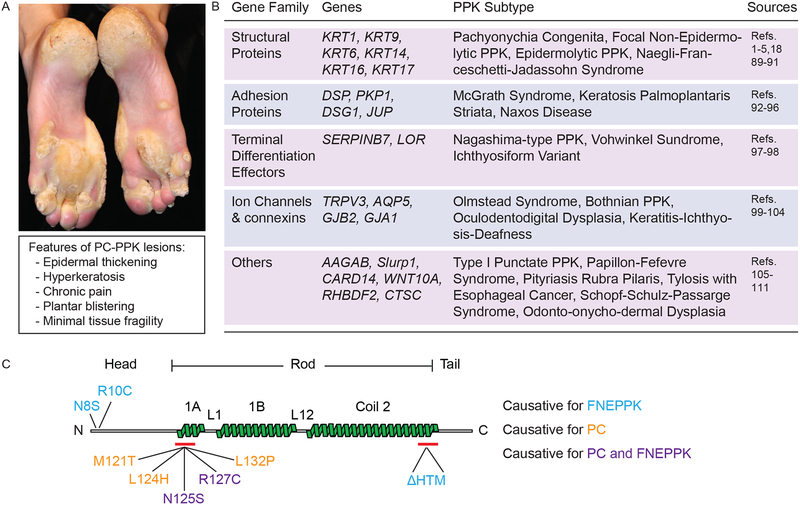
Pachyonychia congenita (PC), a rare genodermatosis, primarily affects ectoderm-derived epithelial appendages and typically includes oral leukokeratosis, nail dystrophy and very painful palmoplantar keratoderma (PPK). PC dramatically impacts quality of life although it does not affect lifespan. PC can arise from mutations in any of the wound-repair-associated keratin genes KRT6A, KRT6B, KRT6C, KRT16 or KRT17. There is no cure for this condition, and current treatment options for PC symptoms are limited and palliative in nature.
This review focuses on recent progress made towards understanding the pathophysiology of PPK lesions, the most prevalent and debilitating of all PC symptoms.
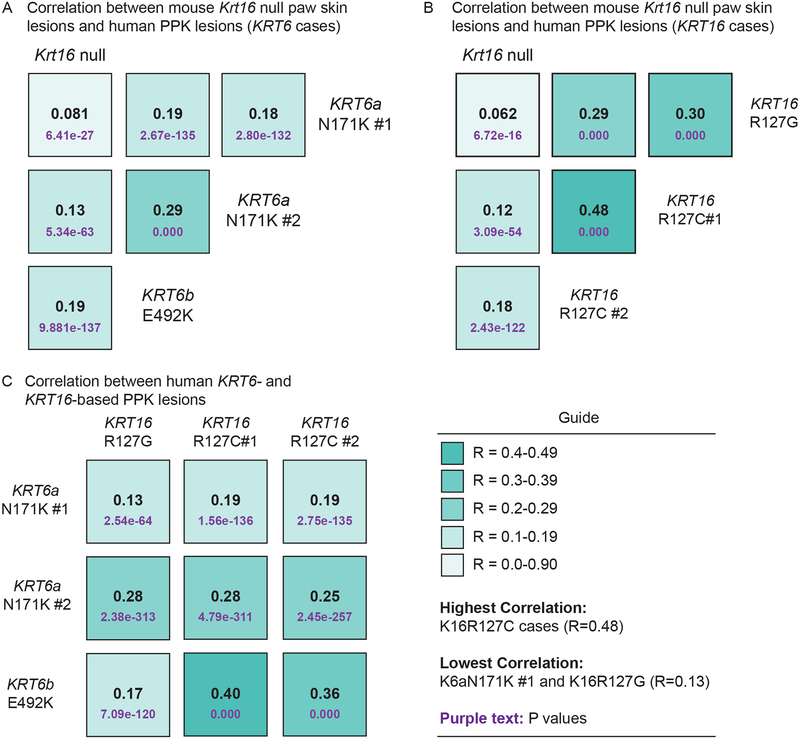
We reviewed the relevant literature with a particular focus on the Krt16 null mouse, which spontaneously develops footpad lesions that mimic several aspects of PC-associated PPK.
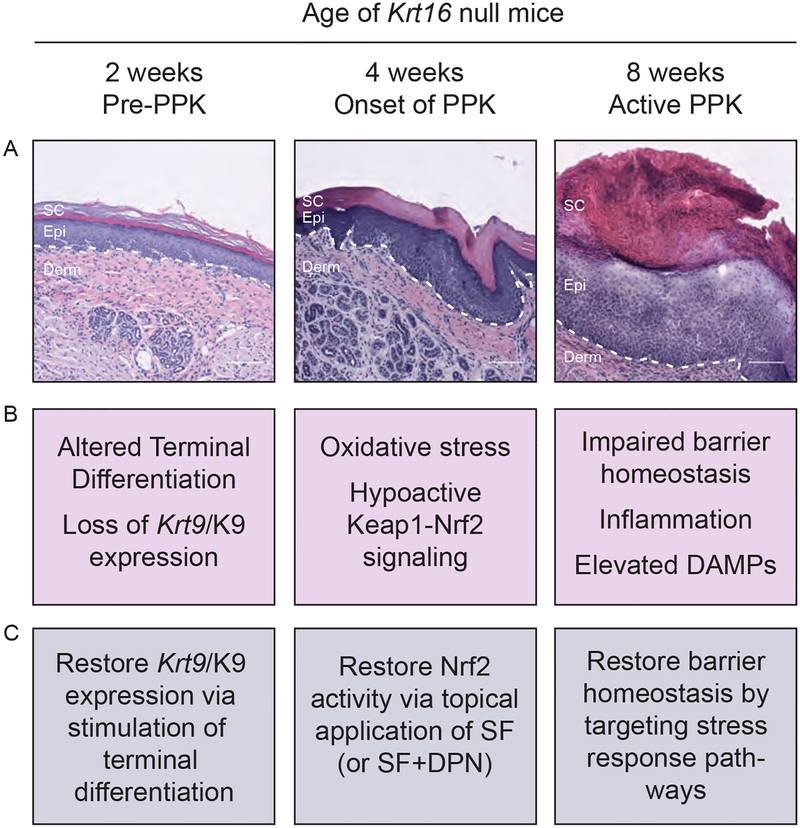
There are three main stages of progression of PPK-like lesions in Krt16 null mice. Ahead of lesion onset, keratinocytes in the palmoplantar (footpad) skin exhibit specific defects in terminal differentiation, including loss of Krt9 expression. At the time of PPK onset, there is elevated oxidative stress and hypoactive Keap1-Nrf2 signalling. During active PPK, there is a profound defect in the ability of the epidermis to maintain or return to normal homeostasis.
The progress made suggests new avenues to explore for the treatment of PC-based PPK and deepens our understanding of the mechanisms controlling skin tissue homeostasis. What's already known about this topic? Pachyonychia congenita (PC) is a rare genodermatosis caused by mutations in KRT6A, KRT6B, KRT6C, KRT16 and KRT17, which are normally expressed in skin appendages and induced following injury. Individuals with PC present with multiple clinical symptoms that usually include thickened and dystrophic nails, palmoplantar keratoderma (PPK), glandular cysts and oral leukokeratosis. The study of PC pathophysiology is made challenging because of its low incidence and high complexity. There is no cure or effective treatment for PC. What does this study add? This text reviews recent progress made when studying the pathophysiology of PPK associated with PC. This recent progress points to new possibilities for devising effective therapeutics that may complement current palliative strategies.
先天性厚甲症(PC)是一种罕见的遗传性皮肤病,主要影响外胚层来源的上皮附属器,通常包括口腔黏膜白斑、甲营养不良和非常疼痛的掌跖角化病(PPK)。尽管PC不影响寿命,但它会严重影响生活质量。PC可能由伤口修复相关角蛋白基因KRT6A、KRT6B、KRT6C、KRT16或KRT17中的任何一个发生突变引起。这种疾病无法治愈,目前针对PC症状的治疗选择有限且本质上是姑息性的。
本综述聚焦于在理解PPK皮损病理生理学方面取得的最新进展,PPK是所有PC症状中最常见且使人衰弱的症状。
我们回顾了相关文献,特别关注Krt16基因敲除小鼠,该小鼠会自发出现模仿PC相关PPK若干方面的足垫皮损。
Krt16基因敲除小鼠中PPK样皮损有三个主要进展阶段。在皮损出现之前,掌跖(足垫)皮肤中的角质形成细胞在终末分化方面表现出特定缺陷,包括Krt9表达缺失。在PPK开始时,氧化应激升高且Keap1-Nrf2信号传导活性降低。在活跃的PPK阶段,表皮维持或恢复正常稳态的能力存在严重缺陷。
所取得的进展为探索基于PC的PPK治疗开辟了新途径,并加深了我们对控制皮肤组织稳态机制的理解。关于该主题已知的信息有哪些?先天性厚甲症(PC)是一种由KRT6A、KRT6B、KRT6C、KRT16和KRT17突变引起的罕见遗传性皮肤病,这些基因通常在皮肤附属器中表达并在损伤后被诱导表达。PC患者有多种临床症状,通常包括指甲增厚和营养不良、掌跖角化病(PPK)、腺囊肿和口腔黏膜白斑。由于PC发病率低且高度复杂,其病理生理学研究具有挑战性。目前尚无治愈或有效治疗PC的方法。本研究增加了什么内容?本文回顾了在研究与PC相关的PPK病理生理学方面取得的最新进展。这一最新进展为设计可能补充当前姑息治疗策略的有效疗法指明了新的可能性。