Department of Cardiology, Peking Union Medical College Hospital, Peking Union Medical College & Chinese Academy of Medical Sciences, Beijing, China.
Institute of Laboratory Animal Sciences, Chinese Academy of Medical Sciences and Comparative Medicine Center, Peking Union Medical College, Beijing, China.
Microbiome. 2019 Apr 26;7(1):68. doi: 10.1186/s40168-019-0683-9.
Coronary artery disease (CAD) is associated with gut microbiota alterations in different populations. Gut microbe-derived metabolites have been proposed as markers of major adverse cardiac events. However, the relationship between the gut microbiome and the different stages of CAD pathophysiology remains to be established by a systematic study.
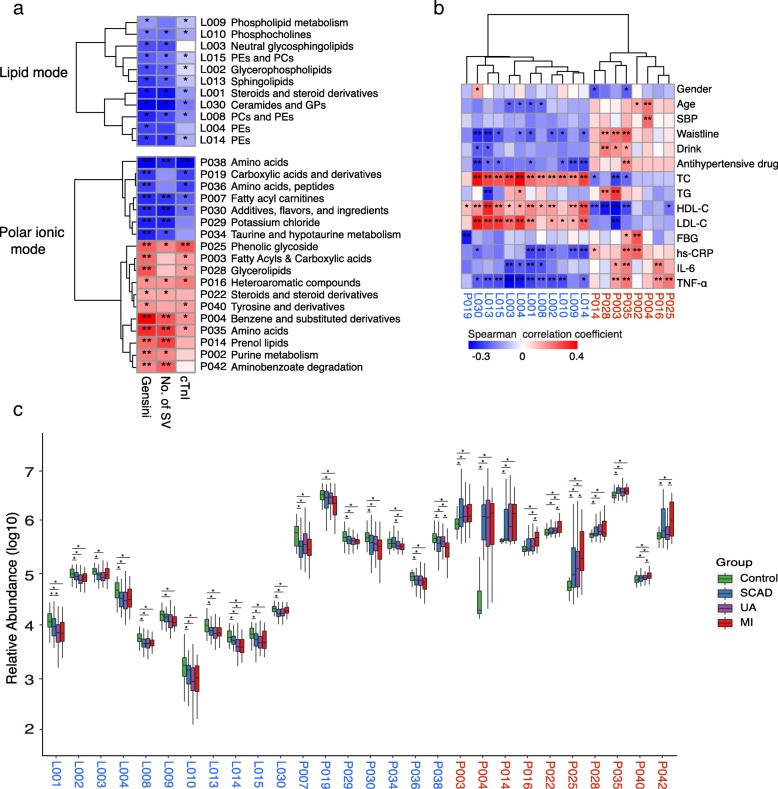
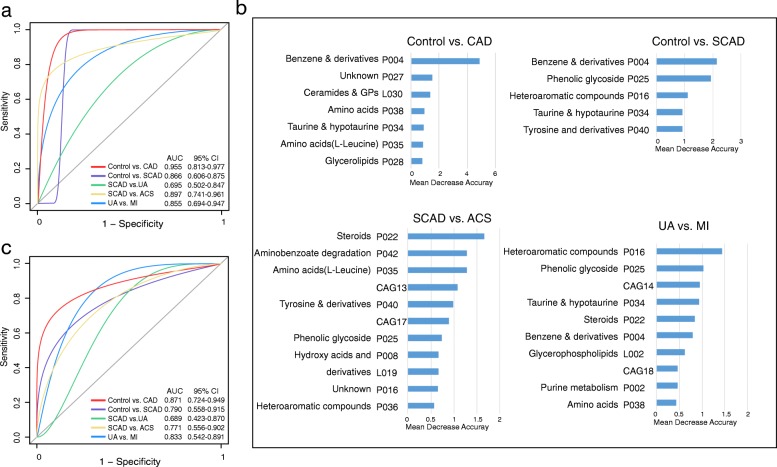
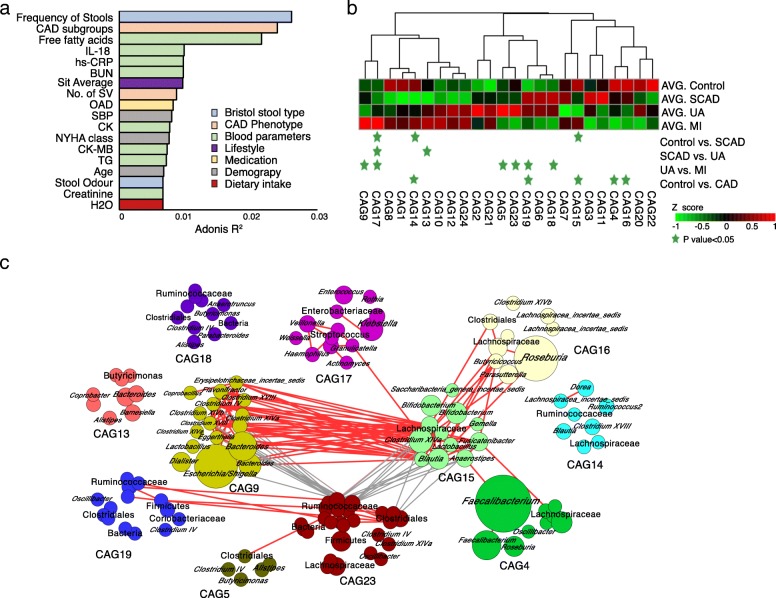
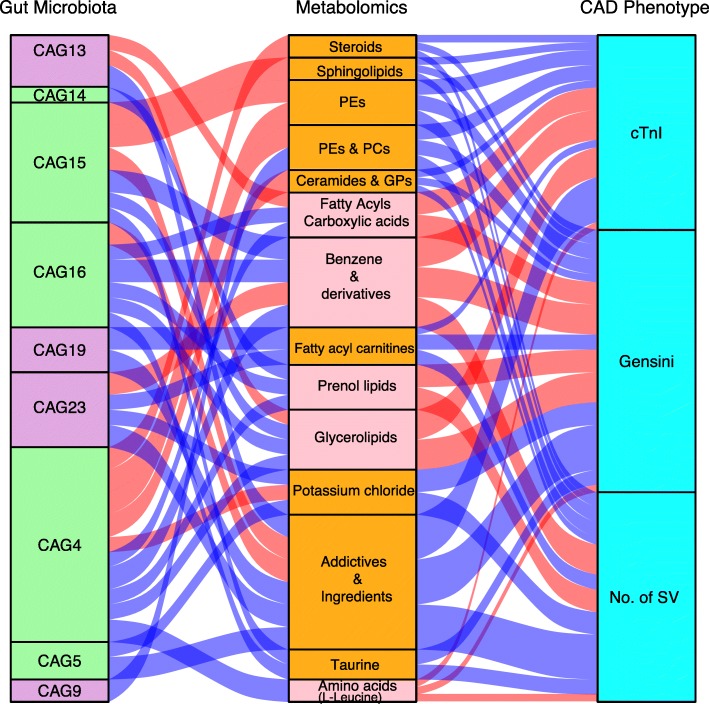
Based on multi-omic analyses (sequencing of the V3-V4 regions of the 16S rRNA gene and metabolomics) of 161 CAD patients and 40 healthy controls, we found that the composition of both the gut microbiota and metabolites changed significantly with CAD severity. We identified 29 metabolite modules that were separately classified as being positively or negatively correlated with CAD phenotypes, and the bacterial co-abundance group (CAG) with characteristic changes at different stages of CAD was represented by Roseburia, Klebsiella, Clostridium IV and Ruminococcaceae. The result revealed that certain bacteria might affect atherosclerosis by modulating the metabolic pathways of the host, such as taurine, sphingolipid and ceramide, and benzene metabolism. Moreover, a disease classifier based on differential levels of microbes and metabolites was constructed to discriminate cases from controls and was even able to distinguish stable coronary artery disease from acute coronary syndrome accurately.
Overall, the composition and functions of the gut microbial community differed from healthy controls to diverse coronary artery disease subtypes. Our study identified the relationships between the features of the gut microbiota and circulating metabolites, providing a new direction for future studies aiming to understand the host-gut microbiota interplay in atherosclerotic pathogenesis.
冠心病(CAD)与不同人群的肠道微生物群改变有关。肠道微生物衍生的代谢物已被提出作为主要不良心脏事件的标志物。然而,肠道微生物组与 CAD 病理生理学的不同阶段之间的关系仍需要通过系统研究来确定。
基于对 161 名 CAD 患者和 40 名健康对照者的多组学分析(16S rRNA 基因 V3-V4 区测序和代谢组学),我们发现肠道微生物群和代谢物的组成都随着 CAD 的严重程度而显著变化。我们确定了 29 个代谢物模块,它们分别被分类为与 CAD 表型呈正相关或负相关,并且在 CAD 不同阶段具有特征性变化的细菌共同丰度群(CAG)由罗氏菌、克雷伯氏菌、梭菌 IV 和真细菌科代表。结果表明,某些细菌可能通过调节宿主的代谢途径,如牛磺酸、鞘脂和神经酰胺以及苯代谢,来影响动脉粥样硬化。此外,构建了基于微生物和代谢物差异水平的疾病分类器,以区分病例和对照者,甚至能够准确区分稳定型冠心病和急性冠状动脉综合征。
总体而言,从健康对照者到不同的冠状动脉疾病亚型,肠道微生物群落的组成和功能都有所不同。我们的研究确定了肠道微生物群特征与循环代谢物之间的关系,为未来旨在了解动脉粥样硬化发病机制中宿主-肠道微生物相互作用的研究提供了新的方向。