Department of Pathology and Immunology, Washington University School of Medicine, Saint Louis, MO, 63110, USA.
Computer Technologies Department, ITMO University, Saint Petersburg, 197101, Russia.
Nat Commun. 2019 May 17;10(1):2209. doi: 10.1038/s41467-019-09990-5.
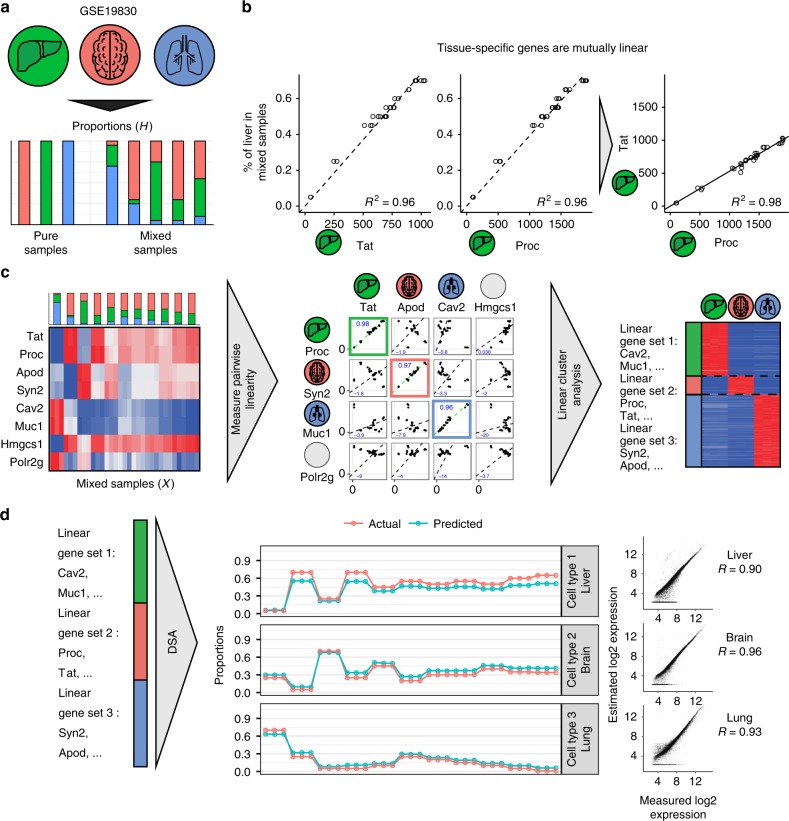
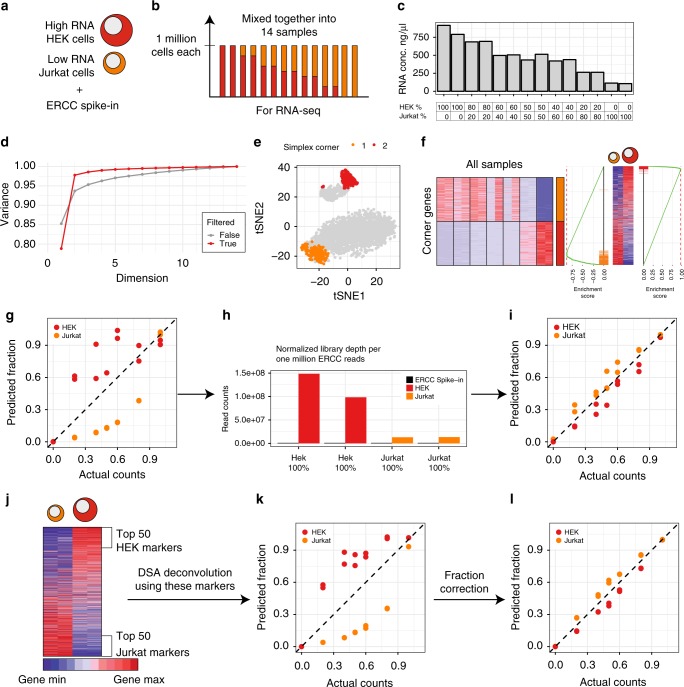
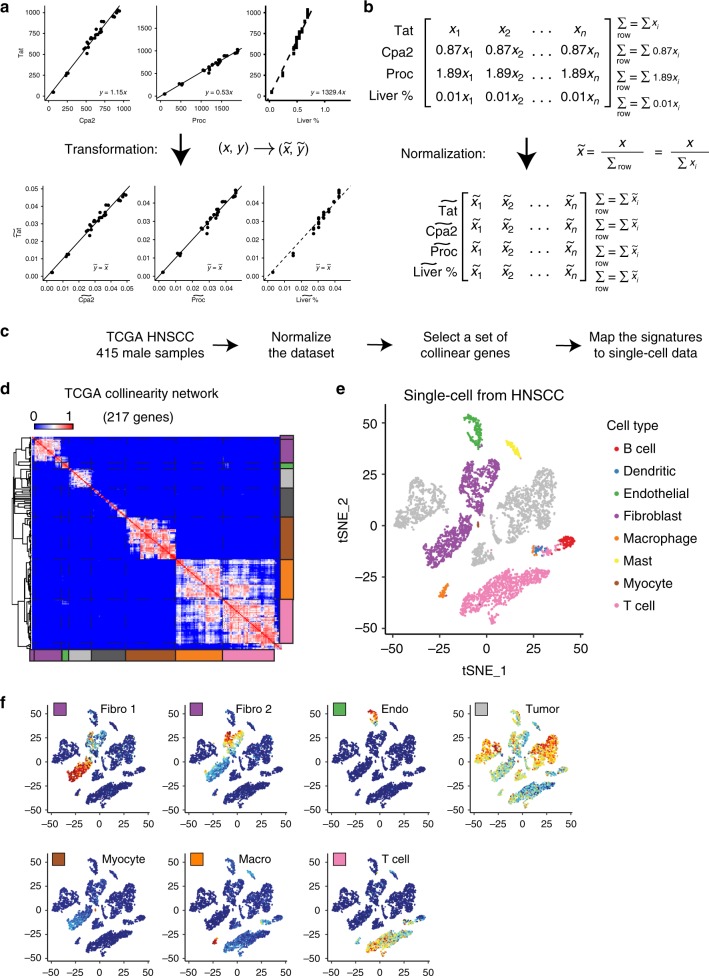
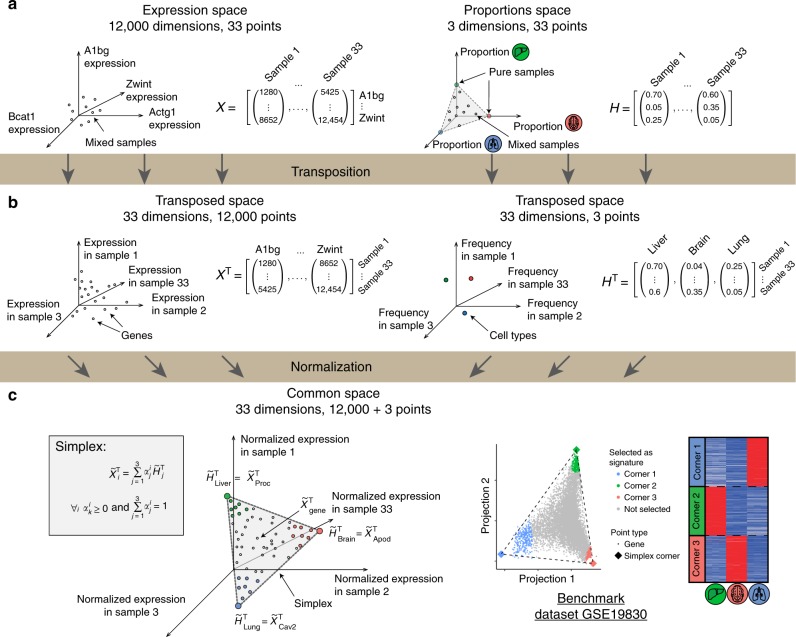
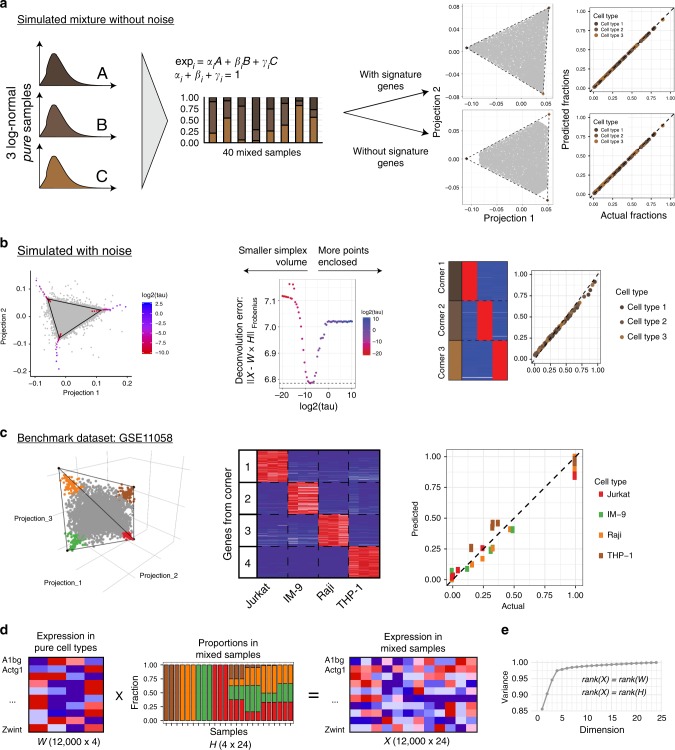
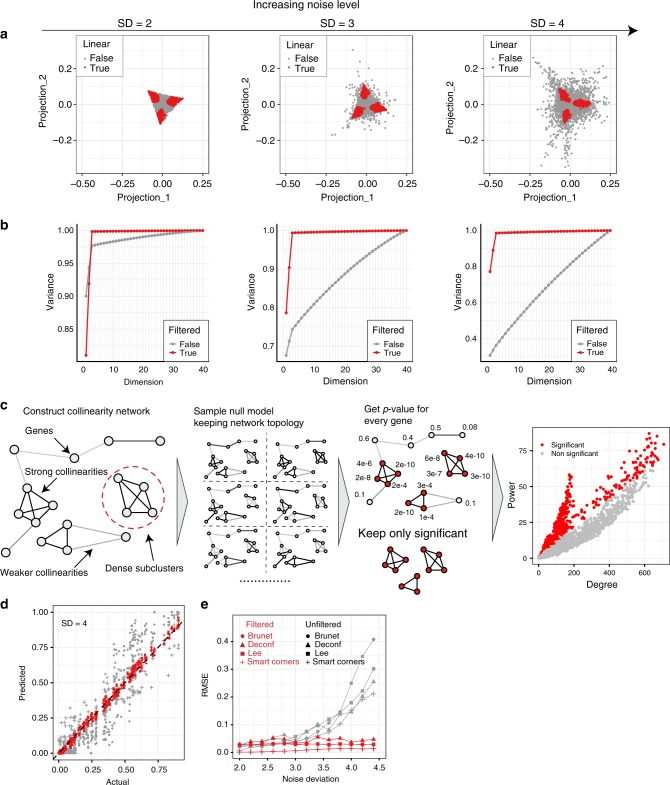
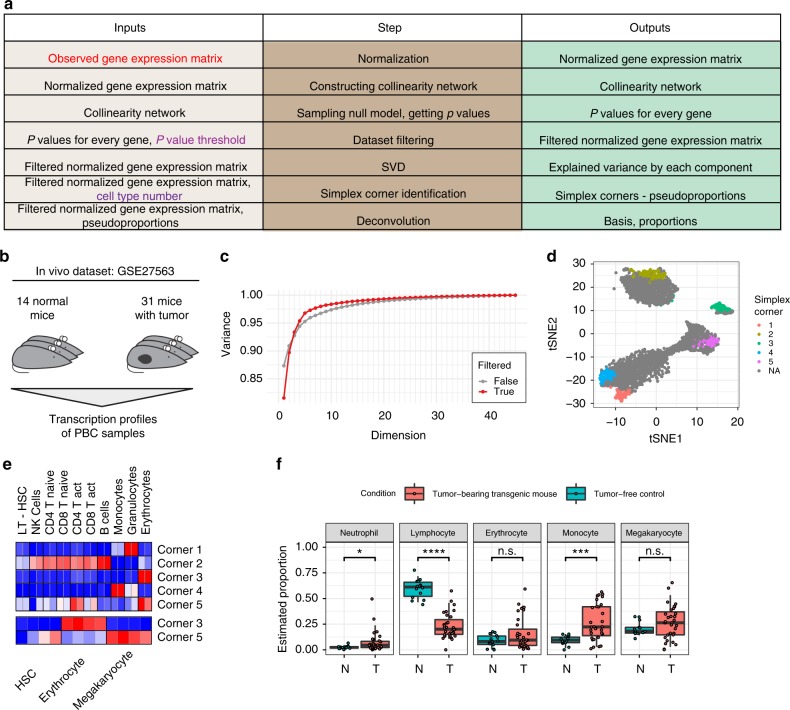
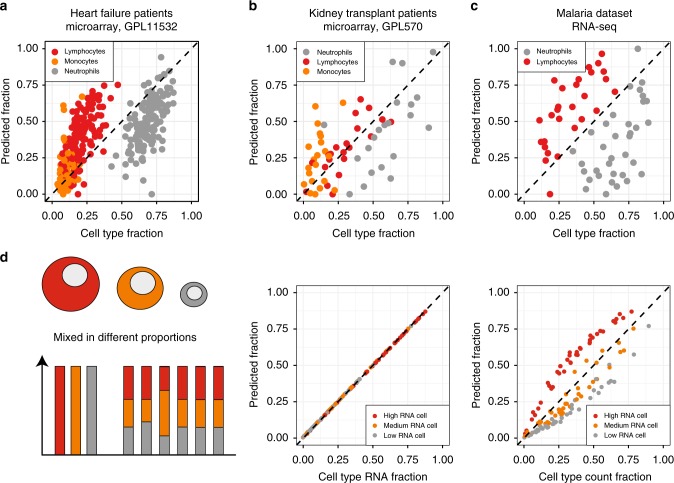
Changes in bulk transcriptional profiles of heterogeneous samples often reflect changes in proportions of individual cell types. Several robust techniques have been developed to dissect the composition of such mixed samples given transcriptional signatures of the pure components or their proportions. These approaches are insufficient, however, in situations when no information about individual mixture components is available. This problem is known as the complete deconvolution problem, where the composition is revealed without any a priori knowledge about cell types and their proportions. Here, we identify a previously unrecognized property of tissue-specific genes - their mutual linearity - and use it to reveal the structure of the topological space of mixed transcriptional profiles and provide a noise-robust approach to the complete deconvolution problem. Furthermore, our analysis reveals systematic bias of all deconvolution techniques due to differences in cell size or RNA-content, and we demonstrate how to address this bias at the experimental design level.
异质样本的整体转录谱变化通常反映了单个细胞类型比例的变化。已经开发了几种强大的技术,可根据纯成分或其比例的转录特征来剖析此类混合样本的组成。然而,在没有关于单个混合物成分的信息的情况下,这些方法是不够的。这个问题被称为完全去卷积问题,在这个问题中,在没有关于细胞类型及其比例的先验知识的情况下,揭示组成。在这里,我们发现组织特异性基因的一个以前未被识别的特性 - 它们的相互线性,并利用它来揭示混合转录谱的拓扑空间结构,并提供一种抗噪的完全去卷积问题的方法。此外,我们的分析揭示了由于细胞大小或 RNA 含量的差异,所有去卷积技术都存在系统偏差,我们展示了如何在实验设计水平上解决这个偏差。