Department of Pediatrics, University of California, San Diego, La Jolla, CA, 92093, USA.
Department of Computer Science & Engineering, University of California, San Diego, La Jolla, CA, 92093, USA.
Nat Commun. 2019 Jun 20;10(1):2719. doi: 10.1038/s41467-019-10656-5.
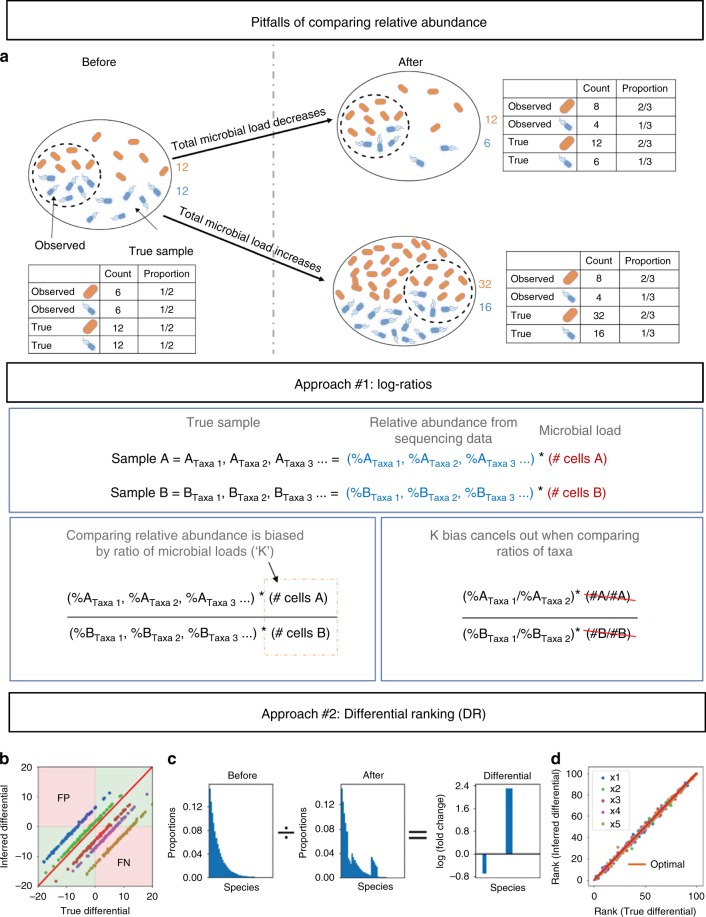
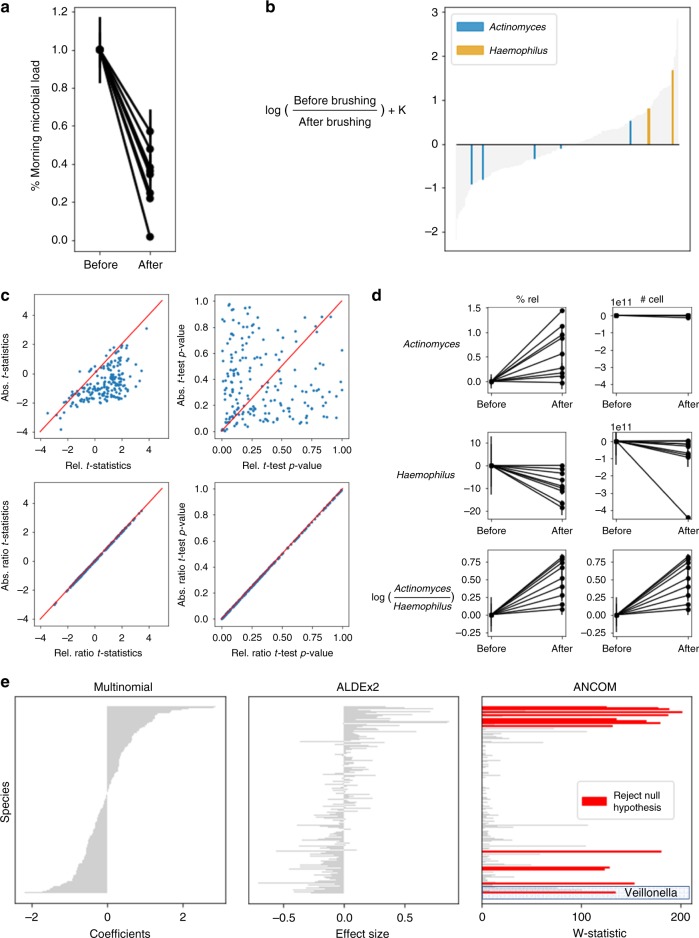
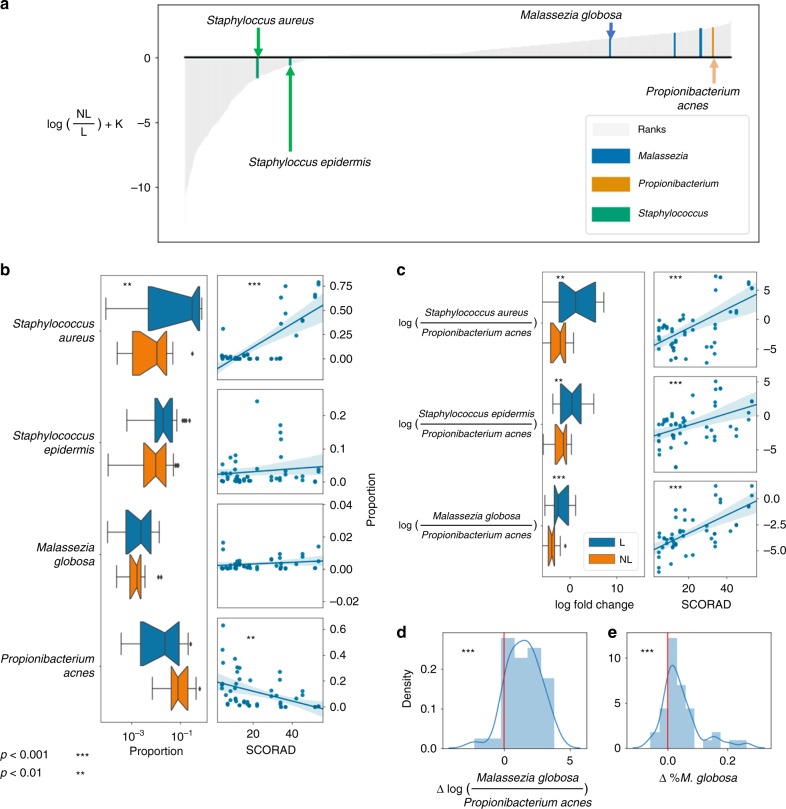
Differential abundance analysis is controversial throughout microbiome research. Gold standard approaches require laborious measurements of total microbial load, or absolute number of microorganisms, to accurately determine taxonomic shifts. Therefore, most studies rely on relative abundance data. Here, we demonstrate common pitfalls in comparing relative abundance across samples and identify two solutions that reveal microbial changes without the need to estimate total microbial load. We define the notion of "reference frames", which provide deep intuition about the compositional nature of microbiome data. In an oral time series experiment, reference frames alleviate false positives and produce consistent results on both raw and cell-count normalized data. Furthermore, reference frames identify consistent, differentially abundant microbes previously undetected in two independent published datasets from subjects with atopic dermatitis. These methods allow reassessment of published relative abundance data to reveal reproducible microbial changes from standard sequencing output without the need for new assays.
差异丰度分析在整个微生物组研究中存在争议。金标准方法需要费力地测量总微生物负荷或微生物的绝对数量,以准确确定分类转变。因此,大多数研究依赖于相对丰度数据。在这里,我们展示了比较样本之间相对丰度的常见陷阱,并确定了两种无需估计总微生物负荷即可揭示微生物变化的解决方案。我们定义了“参考框架”的概念,该概念为微生物组数据的组成性质提供了深刻的直觉。在口腔时间序列实验中,参考框架减轻了假阳性,并在原始数据和细胞计数归一化数据上产生了一致的结果。此外,参考框架识别了以前在特应性皮炎患者的两个独立发表数据集的未检测到的一致、差异丰度微生物。这些方法允许重新评估已发表的相对丰度数据,以从标准测序输出中揭示可重现的微生物变化,而无需新的检测。