Clemson University, Department of Electrical and Computer Engineering, Clemson, SC, 29634, USA.
Clemson University, Department of Genetics and Biochemistry, Clemson, SC, 29634, USA.
Sci Rep. 2019 Jul 5;9(1):9747. doi: 10.1038/s41598-019-46059-1.
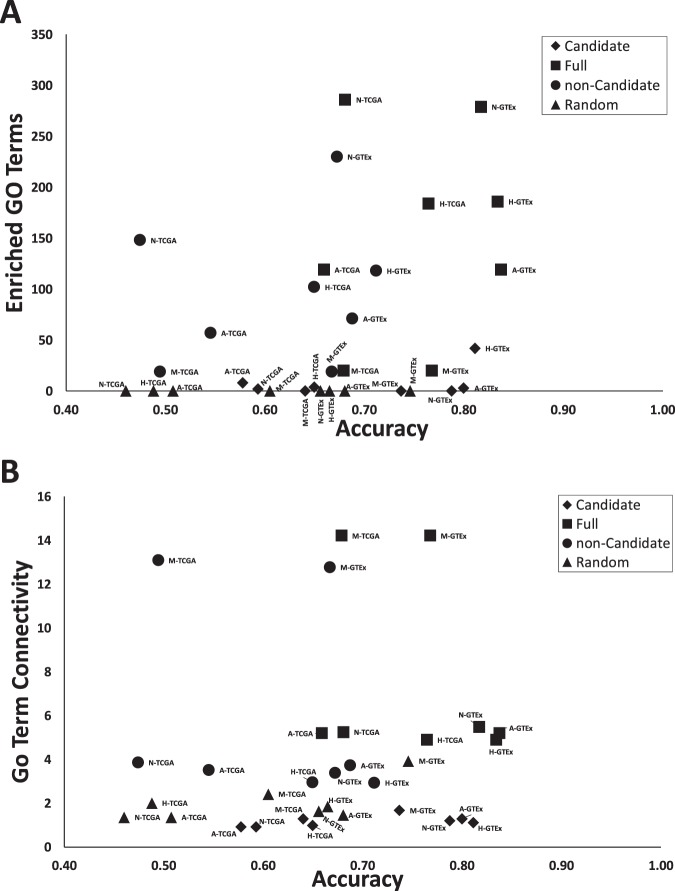
Given the complex relationship between gene expression and phenotypic outcomes, computationally efficient approaches are needed to sift through large high-dimensional datasets in order to identify biologically relevant biomarkers. In this report, we describe a method of identifying the most salient biomarker genes in a dataset, which we call "candidate genes", by evaluating the ability of gene combinations to classify samples from a dataset, which we call "classification potential". Our algorithm, Gene Oracle, uses a neural network to test user defined gene sets for polygenic classification potential and then uses a combinatorial approach to further decompose selected gene sets into candidate and non-candidate biomarker genes. We tested this algorithm on curated gene sets from the Molecular Signatures Database (MSigDB) quantified in RNAseq gene expression matrices obtained from The Cancer Genome Atlas (TCGA) and Genotype-Tissue Expression (GTEx) data repositories. First, we identified which MSigDB Hallmark subsets have significant classification potential for both the TCGA and GTEx datasets. Then, we identified the most discriminatory candidate biomarker genes in each Hallmark gene set and provide evidence that the improved biomarker potential of these genes may be due to reduced functional complexity.
鉴于基因表达和表型结果之间的复杂关系,需要计算效率高的方法来筛选大型高维数据集,以识别具有生物学意义的生物标志物。在本报告中,我们描述了一种通过评估基因组合对数据集样本进行分类的能力(我们称之为“分类潜力”)来识别数据集中最显著的生物标志物基因(我们称之为“候选基因”)的方法。我们的算法 Gene Oracle 使用神经网络来测试用户定义的基因集的多基因分类潜力,然后使用组合方法将选定的基因集进一步分解为候选和非候选生物标志物基因。我们在从癌症基因组图谱 (TCGA) 和基因型组织表达 (GTEx) 数据存储库获得的 RNAseq 基因表达矩阵中量化的来自分子特征数据库 (MSigDB) 的经过策展的基因集中测试了此算法。首先,我们确定了哪些 MSigDB 特征子集对 TCGA 和 GTEx 数据集都具有重要的分类潜力。然后,我们确定了每个特征基因集中最具区分性的候选生物标志物基因,并提供证据表明这些基因的生物标志物潜力提高可能是由于功能复杂性降低所致。